We reviewed retrospectively the radiological findings with CT and MRI of neuroendocrine tumors of our data base. The findings were correlated with pathology results in each of the cases.
We specially go in depth with the ones localized in pancreas, digestive tube and adrenal medulla, since they are the most frequently found in our centre, but we also go through the radiological findings of the ones in unusual localizations
INTRODUCTION
Neuroendocrine tumours are a vast group of neoplasms that arise from neuroendocrine cells located into different organs (Fig 1). It is uncertain but possible that all neuroendocrine cells come from the same embryologic origin, the neural crest.
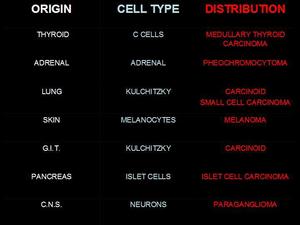
Fig.: Fig 1: Localization of neuroendocrine cells and associated tumors.
Neoplasic neuroendocrine cells are characterized by preservation of their cellular function. These cells have secretory granules that contain amines precursors and keep a decarboxilation function (Amine Precursor Uptake and Decarboxilation: APUD) and can synthesize and secret monoamines and hormonal peptides. The secretion of this hormonal containing is intermittent and sporadic and depends on different factors some of them unknown. Neuroendocrine cells can change the secretion from an hormone to another. The liberation to blood stream of these substances produces typical symptoms according to the hormone produced.
These tumours may be associated with multiple endocrine neoplasia syndrome (MEN I) and phakomatoses.
Tumor diagnosis is based on clinical setting and hormone dosage. CT and MRI are usually the imaging methods used to localize and evaluate tumour extension. If the tumour has a very small size, angiography can help localizing it.
PANCREATIC NEUROENDOCRINE TUMORS
INTRODUCTION
Pancreatic islet cell tumors (PICT) are very rare (5 cases/million inhabitants). PICTs secrete hormones in a variable degree; according to this, they can be classified into syndromic (functioning) and non syndromic (non-hyper-functioning) on the basis of clinical and laboratory findings.
Endocrine pancreatic cells are less than 5% of pancreatic volume and they are grouped up in Langerhans islets. The most frequent three types are: BETA cells (insulin producers), ALPHA cells (glucagon producers) and DELTA cells (somatostatin producers). Other minority cells are: D1 (VIP producers); enterochromaffin cells (they synthesize serotonin); pancreatic polypeptid cells (they synthesize PP that stimulates gastric and intestinal hormones and inhibits intestinal mobility) (Fig 2).

Fig.: Fig 2: PICTs Epidemiology
The endocrine differentation can be confirmed with a Grimelius silver stain or with inmunohistochemical labeling for chromogranin and synaptophysin. Histologically discerning between benignant and malignant is very difficult, so malignancy criteria are based on vascular and adjacent organs invasion. A secondary criterion of malignancy is the size: the larger, the more probable to be malignant.
RADIOLOGIC FINDINGS
The most used diagnostic method is the CT. It is performed after administration of oral water or a low density contrast.
Intravenous contrast in essential, in order to detect the primary tumor, identify its vascular enhancement and diagnose liver metastasis. PICTs are well vascularised tumors and hyperenhanced mostly in arterial phase, the less, are hypodense to pancreatic parenchyma and better detected in portal venous phase or pancreatic phase, because of this, it is so important to acquire images in both arterial and portal phases. In basal MRI, PICTs are usually hyperintense in T2WI and hypointense in T1WI, behaving after gadolinium administration the same way that they do in CT.
PICTs produce metastasis in lymphatic ganglions and liver, as most frequent places. Liver metastasis is usually hypervascular. (Fig 3)

Fig.: Fig 3: PICTs Syndromes.
1-. Syndromic PICTs.
These neoplasms are discovered due to its hormonal production. They are smaller than 3 cm and most cases many hormones are released although one of them is most produced and is responsible of the syndrome. The two most common types of PICTs are Insulinoma and Gastrinoma. Other types are Vipoma, Glucagonoma and Somatostatinoma. (Fig 4)
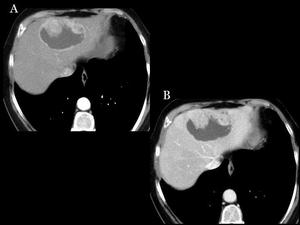
Fig.: Fig 4: Liver metastasis. Non syndromic PICT liver metastasis in CT represented in arterial (A) and portal (B) phases, after pancreatectomy and splenectomy.
1.-Insulinoma
It is the most frequent. Typical signs are: fasting serum glucose levels under 50mg/dl, hypoglycaemia symptoms, and symptomatic relief after glucose administration (triad of Whipple). Some of them are so small that are not possible to be detected by CT/MR so, intraoperative sonography can be useful.
In patients with negative findings but high clinical suspect the selective arterial calcium stimulation and hepatic venous sampling (ASVS) may be used. ASVS sensitivity can be considered at least similar to intraoperative exploration. (Fig 5 & 6)
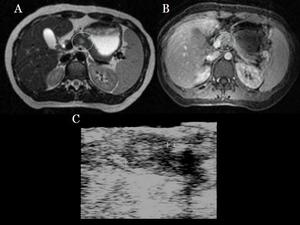
Fig.: Fig 5: Insulinoma: MRI. Hyperintense lesion in pancreatic body in T2WI (A) and after paramagnetic administration in arterial phase (B). Intraoperative sonography (C)
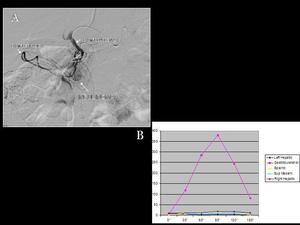
Fig.: Fig 6: Insulinoma. A) A selective artheriography administration on gastroduodenal arthery shows a pancreatic hipervascular mass (insulinoma). B) Selective arterial calcium stimulation on gastroduodenal artery produces and important secretion of insuline which is detected on a hepatic venous sampling. Calcium stimulation on splenic or superior mesenteric arteries does not produce any stimulation.
2.-Gastrinoma
Gastrinomas are usually multiple and have extra pancreatic localization. The called gastrinoma triangle, is the area limited by the pancreas neck and body medially, the second and third portion of duodenum inferiorly and the union of cystic duct, and the common bile duct superiorly.
It is the second most frequent PICT. Its signs are: recurring ulcerous pain and poor response to the treatment; and ulcer unusual localization (post bulbar).
The diagnosis is performed by measuring the levels of gastrina in serum. Gastrinomas are usually malignant, with 30% of liver metastasis when diagnosis is made (Fig. 7 & 8).
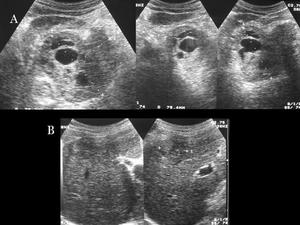
Fig.: Fig 7: Gastrinoma: Sonography. A) Big pancreatic heterogeneous mass, with cystic areas. B) Liver metastasis.
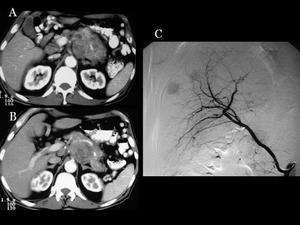
Fig.: Fig 8: Gastrinoma: A & B) CT: Same patient with pancreatic gastrinoma C) Arteriography: Gastrinoma liver metastasis well vascularised.
3.-Glucagonoma
It is called “4D syndrome” because of its clinical findings: diarrhea, dermatosis, depression and deep vein thrombosis. In spite of that, the most frequent clinical manifestation is hyperglycaemia and the typical necrolytic migratory erithema, which is especially localized in genitals, face, abdomen and perineum.
The diagnosis is performed by finding very high levels of glucagon in serum
(more than 1000 pg/ml). These tumors are characterised by a slow growth and a big size at the moment of diagnosis, they are very often malignant and show metastasis in the 75% of cases. Due to their big size (bigger than 5 cm) they usually appear as hyperenhaced lesions with low attenuation areas, cystic lesions are rare.
4.-Vipoma
It is also known as Werner-Morrison Syndrome. These are tumors producer of multiple hormones, including vasoactive intestinal peptide (VIP). VIP stimulates intestinal epithelium, bringing out an important fluid secretion into the lumen, causing a characteristic water diarrhoea.
The diagnosis is performed by observing an increase of VIP concentration in serum with a volume of faeces bigger than a litre per day.
These neoplasias are often malignant and, most cases of VIPomas are situated in the pancreatic tail (75%) and at the moment of diagnosis, show metastasis in a 60% of cases
5.-Somatostatinoma
Diagnosis is made by clinical manifestations and messuring somatostatina level in serum. They are often a big, unique mass and, at the moment of diagnosis, show metastasis in a 50% of cases.
Somatostinoma appears more frecuently in pancreatic head, less frecuently, next to Vater’s ampulla when seats in duodenum (producing obstruction).
2-.Non syndromic PICTs
These are half of all PICTs. They are larger than syndromic PICT when diagnosis is made (usually more than 5 centimetres), manifesting for their compressive symptomatology, or if smaller, detected as a casual image finding.
These PICTs do not release detectable hormones, but secret different substances, probably without any hormonal function.
Non syndromic PICTs are frecuently malignant, due to their size, and in many cases appear in CT with hepatic and ganglionar metastasis as we show in some of our cases. They usually present necrosis, calcification and cystic areas. (Fig 9, 10, 11, 12, 13 & 14)
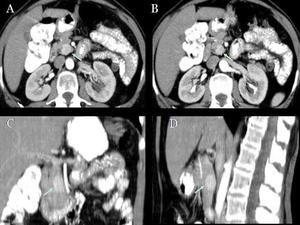
Fig.: Fig 9: Non syndromic PICT in pancreatic head: CT. CT in arterial (A) and portal (B) phases, where a tumoral enhacement in both is observed, being more intense in arterial phase. Coronal (C) and sagital (D) reconstruction
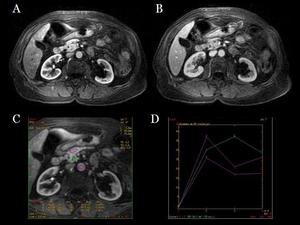
Fig.: Fig 10: Non syndromic PICT in pancreatic head: MR. MR of same patient. T1WI after paramagnetic contrast administration, in arterial (A) and portal (B) phases show an enhacement of the tumoral lesion more intense in the arterial phase. Enhancement curves are shown (c and d) by placing ROIs in pancreatic parenchyma (1), tumor (2) and aorta (3).
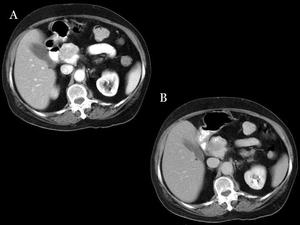
Fig.: Fig 11: Non syndromic PICT. Pancreatic head PICT with important enhancement after contrast administration shown in arterial and portal phases. (A and B)
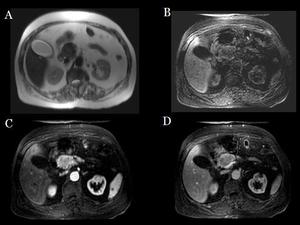
Fig.: Fig 12: Non syndromic PICT: MRI. Hypointense lesion in pancreatic head in T2WI (A), hypointense in FAT SAT T1WI (B), and after paramagnetic contrast administration in arterial (C) and portal (D) phases it is more intense in arterial phase.
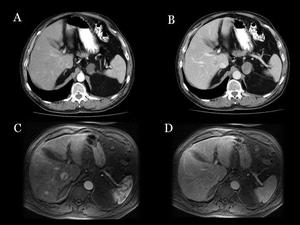
Fig.: Fig 13: Liver metastasis in non syndromic PICT. Liver lesions were not well seen in dual phase CT ( A and B). Days after, MR after gadolinium administration, shows multiple liver metastasis (C: arterial phase, D: portal phase)
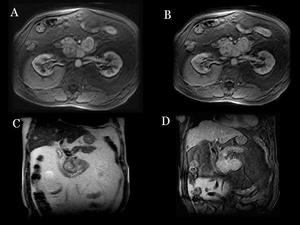
Fig.: Fig 14: Non syndromic PICT:MRI. Same patient after contrast administration in T1WI arterial (A) and portal(B) phases, and coronal images (C y D) .
INTESTINAL CARCINOID TUMORS
INTRODUCTION
Carcinoid tumors arise from neuroendocrine cells located in the intestinal mucosa or submucosa, they have slow growth and spread to the mesentery (40 – 80 %) by direct extension or lymphatic spread.
Intestinal carcinoid are rare, just 5,5 % of the tumors of the gastrointestinal tract. They can be active and secret hormones, more frequently in patients with hepatic metastases. The 40 % of gastrointestinal carcinoids are multiple.
The symptoms are unspecific like abdominal pain and obstruction or carcinoid syndrome (diarrhoea, flushing and bronchial constriction).
The malignancy of carcinoid tumors is based on the invasion of different structures or evidencing metastases, rather than the histologic appearance. The incidence of metastases is correlated with the tumor size: metastases are more frequent for tumor size of 2 cm or higher.
RADIOLOGIC FINDINGS
The most frequent barium studies appearance is as solitary or multiple submucosa nodules with thickening of the wall, when it extends through the muscular layer. If the carcinoid extends outside the intestinal wall it infiltrates the mesentery and appears as a mass effect with fixation of the small bowel loops (Fig 15 and 16).
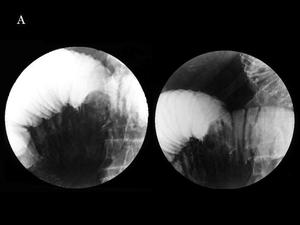
Fig.: Fig. 15 & 16: Intestinal carcinoid with mesenteric spread. A) Barium study. Submucosal nodule with mucosal folds’ retraction at the mesenteric edge.
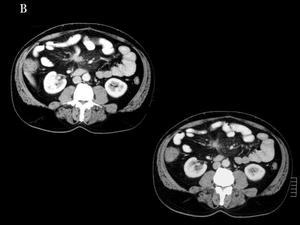
Fig.: Fig. 15 & 16: Intestinal carcinoid with mesenteric spread. B) Contrast-enhanced abdominal CT. Small spiculated nodule in the mesenteric fat.
On CT the intestinal nodules are small so they are difficult to be detected and the mesenteric tumor is the dominant imaging finding. Administration of water and performing an arterial and venous phases contrast-enhanced study may help the detection of primary focus. The mesenteric extension is better detected as a soft enhancing mass with linear bands radiating into the mesenteric fat (which represents fibrotic proliferation and desmoplastic reaction). Calcification into the mass (70 %) and thickening of adjacent small bowel loops (that represents tumor infiltration or ischemia because of encasement of mesenteric vessels) may be seen (Fig 17).
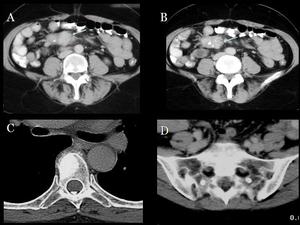
Fig.: Fig 17: Intestinal carcinoid with mesenteric spread and bone metastases. A, B) Contrast-enhanced abdominal CT. Spiculated mesenteric mass with some calcifications, corresponding to mesenteric dissemination from an primary intestinal carcinoid. C,D) Multiple blastic metastases in a thoracic vertebra and sacrum.
Carcinoid metastases are usually in the liver, lung, bone or peritoneum. Liver metastases are hypervascular lesions so they enhance intensely on arterial phase (Fig 18).
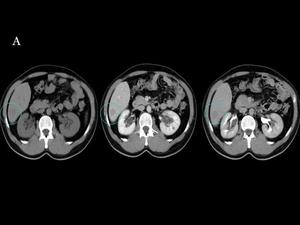
Fig.: Fig 18 & 19: Liver metastasis from a intestinal carcinoid. A) Unenhanced and contrast-enhanced abdominal CT shows a nonenhancing hypoattenuating hepatic nodule both in the portal and delayed phase.
The MRI appearance is like an intestinal mass with varied signal on T1WI and T2WI that enhances with gadolinium. Sometimes the mass is not seen but instead we can see bowel wall thickening, with enhancement after IV contrast agent administration that represents submucosal nodules. The mesenteric extension appears like a homogeneous mass with intense enhancement. The hepatic metastases are hypointense on T1WI, hyperintense on T2WI and hypervascular on arterial phase becoming isointense on portal venous phase, sometimes they have central necrosis with peripheral enhancement (Fig 19).
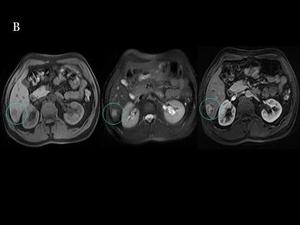
Fig.: Fig 18 & 19: Liver metastasis from a intestinal carcinoid. B) The liver nodule shows a low signal on unenhanced T1WI and a high signal on T2WI. After gadolinium injection, the lesion shows an avid enhancement in the arterial phase, consistent with a hypervascular metastasis.
The angiography is used for localization of primary tumor because is well vascularised.
NEUROENDOCRINE TUMORS OF THE LUNG
INTRODUCTION
Neuroendocrine tumors of the lung arise from Kulchitzky cells (chromaffin cells) of the bronchial mucosa. The latest classification of pulmonary neuroendocrine tumors, proposed in 1991 by Travis et al includes four types: typical carcinoid, atypical carcinoid, large-cell neuroendocrine carcinoma (LCNC) and small-cell lung cancer (SCLC). The differenciation between these tumors is based on their histopathologic features (figs 20 and 21): carcinoid tumors consist of small nests of uniform neuroendocrine cells separated by prominent vascular stroma. Atypical carcinoids show more mitoses than typical carcinoids do; moreover, areas of necrosis usually absent in typical carcinoids, are not uncommon in atypical ones. Calcification and even ossification (more frequent in typical carcinoids) are seen in up to 30% of these tumors. LCNCs, an intermediate category between carcinoids and small-cell carcinoma, both in terms of histopathology and prognosis, show a high mitotic rate and extensive areas of necrosis. SCLC, which has the poorest prognosis of all lung cancers, has high cellularity with a very high mitotic rate, with a poorly preserved architecture of tumor clusters.
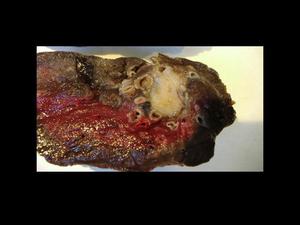
Fig.: Fig 20: Lung carcinoid. Macroscopic specimen.
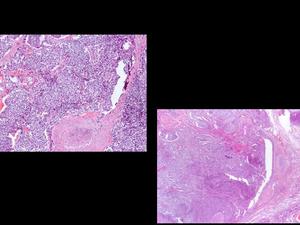
Fig.: Fig 21: Lung carcinoid. Histopathology.
Typical carcinoids are not related to smoking habit, whereas the rest of types are more frequent in smokers, especially SCLC. The mean age of patients affected by bronchial carcinoid range from 40 to 60 years, 60 to 70 years for LCNT, and 70 to 80 for SCLC.
RADIOLOGIC FINDINGS
Carcinoid tumors are centrally located masses, arising in 80% of cases from the main, lobar or segmental bronchus wall. The typical image is a hilar or perihilar nodule or mass. However, peripheral nodules are not a rare form of presentation. The tumors are well-defined masses, with a round or lobulated shape (fig 22) and they rarely show peripheral spiculation. They tend to have a homogeneous density on CT scans and, as they are highly vascular tumors, they show intense contrast enhancement, although this is not a constant feature. Eccentric calcifications are common and they are easily identified on CT, but rarely in conventional radiographs. Other typical findings are the consequences of bronchial obstruction, such as air trapping, atelectasis, postobstructive pneumonia and mucus plugging of peripheral bronchi. Both typical and atypical carcinoid may be associated with hilar or mediastinal lymphadenoadenopathy, which occur more frequently from atypical carcinoids.
Distant metastases are found in up to 15% of bronchial carcinoids, and they involve the liver, adrenal glands, bone and brain (fig 23, 24 and 25) There are no radiologic findings specific enough to distinguish a typical from an atypical carcinoid, although atypical ones tend to have an increased frequency of thoracic lymphadenopathy and metastases at the moment of presentation.
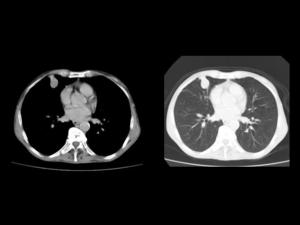
Fig.: Fig 22: Lung carcinoid. Unenhanced chest CT: Well-defined nodule in the middle lobe, in a peripheral location, that reaches the pleura, which is thickened. It has a soft tissue density and no calcifications are observed. This is a quite unspecific appearance for a lung primary tumor.
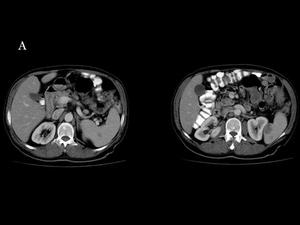
Fig.: Fig 23, 24 & 25 : Metastases from a lung carcinoid. Contrast-enhanced body CT. A) Spleen metastases: Hypoattenuating nodules in the spleen.
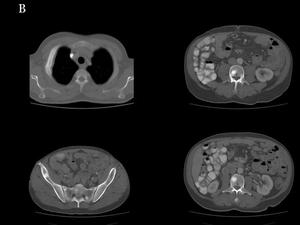
Fig.: Fig 23, 24 & 25 : Metastases from a lung carcinoid. Contrast-enhanced body CT. B) Bone metastases: Multiple blastic lesions in the lumbar spine, sacrum and iliac wings. In a right rib it is seen an expanding lesion associated with a soft-tissue mass adjacent to the pleura.
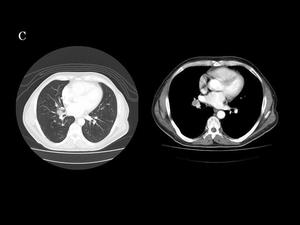
Fig.: Fig 23, 24 & 25 : Metastases from a lung carcinoid. Contrast-enhanced body CT. C) Lung metastases: Multiple nodular metastases in both lungs. Right hilar lymphadenopathy is also seen, with a hypoattenuating component. Primary carcinoid tumor was located in the middle lobe.
LCNCs are rare tumors whose radiologic findings are non-specific and similar to other non-small cell lung carcinomas.
SCLCs account for 20% of bronchogenic carcinomas. Approximately 90-95% of SCLCs occur centrally (fig 26), arising in the main or lobar bronchus. At the time of diagnosis, patients usually have extensive disease with rapid tumor growth. The hallmark of this tumor’s imaging in the presence of extensive mediastinal or hilar lymphadenopathy, (even without an identifiable primary tumor), with displacing or narrowing of the tracheobronchial tree or major mediastinal vessels. Intratumoral calcifications are seen in up to 23% of SCLCs. Its enhancement pattern is similar to those of non small-cell lung carcinomas.
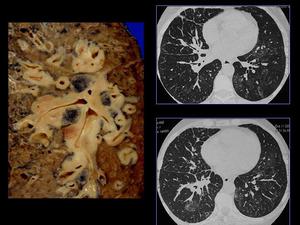
Fig.: Fig 26: Small cell lung carcinoma. Macroscopic specimen and CT: peribronchial spread.
PARAGANGLIOMAS
INTRODUCTION
A single collection of neuroendocrine tissue is called a paraganglion and the entire chain of tissue constitutes the paraganglia. They arise from neural crest progenitor cells. Paraganglia in the head and neck migrate along a branchiomeric distribution, whereas those in the chest, abdomen, and pelvis follow the parasympathetic nerve fibers along the perivertebral-periaortic axis. The most tipical localitation of paraganglia tumors is the adrenal medulla and it is known as pheochromocytoma. Extraadrenal paraganglia, defined as paraganglia located outside the adrenal gland, can be divided in two broad groups: paraganglia associated with the parasympathetic system and those related to the sympathetic system. In addition, small paraganglia are found within viscera such as the urinary bladder and gallbladder.
Some paragangliomas are hormonally active with excess catecholamine secretion. They are rare in the head and neck region but occur frequently in the thorax and abdomen. The clinical presentations related to catecholamine hypersecretion include headache, palpitations, and sweating. For those patients with nonfunctioning paragangliomas, the clinical manifestation is a palpable mass or pain related to the localization of the tumor mass. In addition, approximately 10% of paragangliomas are clinically silent. The term glomus tumor was used to describe the rich arborisation of blood vessels and nerves seen in these masses
PHEOCHROMOCYTOMAS
Pheochromocytomas are a catechohamine –secreting tumor. 98% of pheochromocytomas are located in the abdomen and 90% in the adrenal glands. Clinically there are two different behaviours: if the tumor is non functioning it is generally a large asymptomatic tumor. The hyperfunctioning tumors can produce hypertension, headache, anxiety, palpitation and tachycardia because of catecholamine secretion and they can be life threatening.
Most are unilateral. 10% are bilateral, 10% are associated with inheritable diseases and 10% of them are malignant and can metastasize.
On CT generally they are high attenuation mass (more than 10 UH) but sometimes they have lipid degeneration and it is seen like a soft tissue mass with fat. After IV contrast administration they have heterogeneous enhancement (because of their cystic changes) and have variable washout patterns.
On MRI they are hypointense on T1WI, hyperintense on T2WI and frequently with intratumoral cystic areas. They have hyperenhance after gadolinium administration. When there is fat or hemorrhage into the mass they produce high signal on T1WI images (Fig 27).
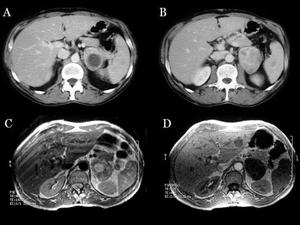
Fig.: Fig 27: Pheochromocytoma. A, B) Contrast-enhanced abdominal CT shows a left adrenal mass with a heterogeneous enhancement with a hypoattenuating component consistent with necrotic or cystic changes. C, D) Abdominal MRI. The tumor is predominantly hyperintense on T2WI and hypointense on unenhanced T1WI.
PARAGANGLIOMAS OF THE HEAD AND NECK
Paragangliomas of the head and neck are located within the parasympathetic nervous system, and the four most common sites are the carotid body at the common carotid artery (CCA), the jugular foramen, along the vagus nerve, and within the middle ear.
CAROTID BODY PARAGANGLIOMAS
The carotid body tumor is the most common paraganglioma of the head and neck. It is most frecuently situated on the posteromedial wall at the bifurcation but does not narrow the calibre of the ECA (external carotid artery) and ICA (intern carotid artery)
The CT appearance is a well-defined soft- tissue mass with homogeneous and intense enhancement because of their hypervascularity. They are rarely inhomogeneous areas due to focal thrombi or hemorrhage. With disease progression, the lesion may extend to the skull base and the parapharynx spaces.
On MRI, typically have low intensity signal on T1WI and high signal on T2WI (Fig 28). A characteristic appearance of this lesion are multiple punctuate areas of signal void on all MRI sequences and adjacent hyperintense foci (due to haemorrhage), this has been described as “salt-and-pepper sign” but it is not specific (Fig 29).
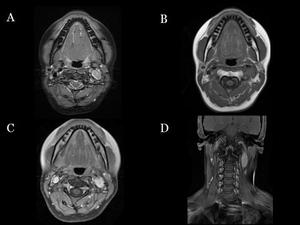
Fig.: Fig 28: Left carotid glomus. Neck MRI. A) Axial fat-suppressed T2WI. Hyperintense mass on the left carotid bifurcation. B) On T1WI the lesion is hypointense. C, D) Axial and coronal contrast-enhanced T1WI shows marked gadolinium enhancement.
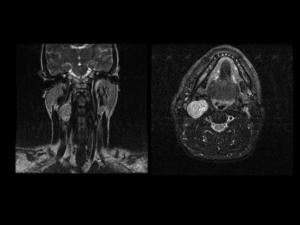
Fig.: Fig 29: Glomus. Coronal and axial IR MRI that shows a mass on carotid bifurcation hyperintense with punctuates areas of signal void due to hemorrage described as “salt and pepper sign”.
The angiographic appearance of a paraganglioma is a hypervascular mass with enlarged feeding arteries (the most common are the ascending pharyngeal artery and the ascending cervical artery) tumor blush and rapidly draining veins. Angiography is used for diagnosis, preoperative embolisation or palliative therapy (Fig 30).
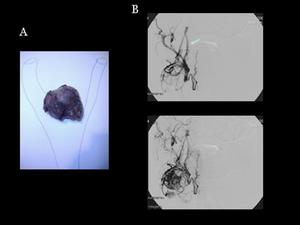
Fig.: Fig 30: Carotid glomus: A) Macroscopic specimen. B) Glomus ateriography: IV contrast administration on ECA shows a hypervascular mass in carotid bifurcation with enlarged feeding ascending pharyngeal artery.
GLOMUS JUGULARE TUMORS
Glomus jugulare tumours arise in the jugular foramen from Jacobson's nerve or Arnold's nerve. Patients may present with pulsatile tinnitus, hearing loss, or vertigo.
CT findings of glomus jugulare tumours are enlargement of the jugular foramen and irregular margins.
NEUROENDOCRINE TUMORS IN NEUROFIBROMATOSIS TYPE 1 Neurofibromatosis type 1 (NF-1) has an autosomal dominant inheritance pattern. It results from a mutation of a tumor-suppressor gene, located in the large arm of chromosome 17. In about 50% of cases, the disease is due to a sporadic mutation, and the rest of cases are familiar. General clinical features are café au lait spots, neurofibromas, Lisch nodules (melanocytic hamartomas of the iris), axillary or inguinal freckling, bone dysplasias and a wide spectrum of benign and malignant neoplasms.
Neuroendocrine tumors are more frequent in patients with NF-1 than in the general population (fig 31). The most frequent ones are intestinal carcinoids and pheochromocytomas.
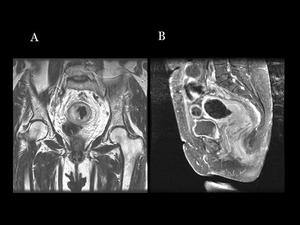
Fig.: Fig 31: Neuroendocrine rectal tumor in a patient with neurofibromatosis type 1. A) Coronal T2WI that shows an eccentric thickening of the rectal wall with a high signal in this sequence B) Gadolinium-enhanced fat-suppressed sagittal T1WI shows diffuse enhancement of the tumor.
Intestinal carcinoids in NF-1 arise typically in or near the ampulla of Vater. They may present with symptoms of biliary obstruction, and therefore raise the suspicion of a pancreatic or bile duct tumor. Pancreatic adenocarcinoma is commonly a hypoattenuating and infiltrating mass, whereas a periampullary carcinoid has an intramural or polypoid configuration, and tends to be focal. It is however very difficult to differenciate these tumors from other periampullary neoplasms.
Pheochromocytoma in NF-1 patients is frequently symptomatic. There is a curious association of pheochromocytomas and duodenal carcinoids in patients with NF-1.
NEUROENDOCRINE TUMORS IN VON HIPPEL-LANDAU DISEASE
Von Hippel-Lindau (VHL) disease is a rare autosomal dominant familial syndrome associated with brain, retinal and spinal cord hemangioblastomas, renal cysts and renal cell carcinoma, pancreatic cysts and two types of neuroendocrine tumors: pheochromocytoma and pancreatic islet cell tumors.
Pheochromocytoma is by far the most frequent neuroendocrine tumor in VHL. They are bilateral in up to 50% of patients and are usually benign (although some of them can metastatize). Most tumors are localized in the adrenal glands, but 15-18% of the lesions are found in an extraadrenal location (paragangliomas). They are usually non-symptomatic and do not have elevated serum catecholamine levels.
Pancreatic neuroendocrine tumors (figs 32 and 33) have a low prevalence in VHL disease. They are usually non-functioning and asymptomatic, are often multiple, have a slow rate of growth and have non particular pancreatic location. They don’t seem to be related with pancreatic cystic disease.
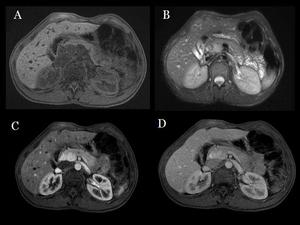
Fig.: Fig 32 & 33: Islet-cell tumors in Von Hippel-Lindau disease: Two lesions in the head of the pancreas show low signal in T1WI (A), high signal in T2WI (B), and intense gadolinium enhacement in the arterial phase(C), and become isointense in the portal phase(D).
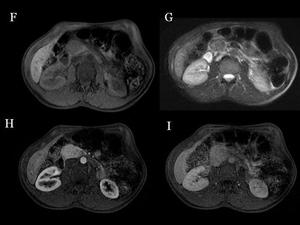
Fig.: Fig 32 & 33: Islet-cell tumors in Von Hippel-Lindau disease: Another lesion is hypointense to pancreatic tissue in T1WI (F) and also in T2WI (G). Enhancing characteristics (H, arterial phase, I, delayed phase) are similar to the other two lesions.
Interestingly, pancreatic neuroendocrine tumors often coexist with pheochromocytoma in VHL patients. The association between these two tumors has arisen some speculations about a possible continuum of VHL disease and multiple endocrine neoplasia (MEN) syndromes.
NEUROENDOCRINE TUMORS IN MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
Multiple Endocrine Neoplasia (MEN) comprises various genetically determined disorders with a predisposition to tumor development within two or more components of the endocrine system. There are two major forms: MEN-1 and MEN-2
MEN-1 is an autosomal dominant condition with a high penetrance. Parathyroid tumors, pancreatic islet cell tumors and pituitary tumors are the major components of the disease. Facial angiofibromas, collagenomas, adrenal cortical tumors and lipomas are also frequent.
The most useful radiologic technique to detect parathyroid adenomas is MRI. Parathyroid adenomas have an elevated signal intensity on T2WI and STIR sequences and a reduced signal on T1WI.
Most pancreatic tumors in MEN-1 (fig 34) are functioning, with less malignant potential than sporadic pancreatic tumors. They tend to be small (less than 2 cm) and may be multiple; a larger size and the presence of calcifications suggest malignancy.
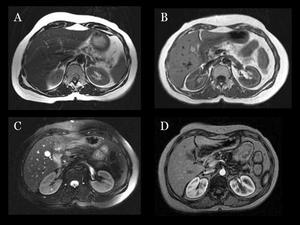
Fig.: Fig 34: MEN-1 syndrome: A) Axial T1WI: Bilateral adrenal hyperplasia. B) Small nodular lesion in the head of the pancreas, hypointense in this T1-weighted axial image. C) The lesion shows a high signal on T2WI. D) Dynamic gadolinium-enhanced out-of-phase T1WI shows an avid contrast uptake in the arterial phase. This lesion proved to be histologically a neuroendocrine pancreatic neoplasia.
MEN-2 is also an autosomal dominant cancer syndrome that is characterized by the association of medullary thyroid carcinoma (MTC), that is present in 100% of cases, and pheochromcytoma in 50% of cases. Two subtypes are recognized: MEN-2A (with parathyroid hyperplasia or tumors) and MEN-2B (with a characteristic marfanoid habitus and mucosal neuromas).
At CT and MRI, MTC is solid and may contain calcifications. Local, nodal and distant spread can be readily identified.
