CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD,
defined on the basis of spirometric evidence of airway obstruction,
encompasses several distinct but overlapping obstructive syndromes not entirely reversible,
including emphysema,
chronic bronchitis,
and reduction in the caliber and number of small airways in the lung.
Bronchiolitis is the earliest lesion in COPD.
Emphysema is formed distal to surviving bronchioles,
supported by collateral ventilation of acini within the same lobule that lost terminal bronchioles.
Bronchitis or inflammation of the large airways is secondary to an innate immune response to the inhaled toxic particles and gases,
mediated by various populations of T-cells,
macrophages,
and neutrophils that lead to hypersecretion of mucus from goblet cells,
thickening,
inflammation,
and fibrosis of the bronchial and bronchiolar walls.
This leads to a further reduction in luminal diameter and predisposition toward expiratory collapse of both the large and small airways.
On imaging,
the walls of the large airways will be thickened and endobronchial mucus plugging may be visualized (Fig.
1).
Bronchial wall thickening is an important independent predictor of FEV1 and of the risk of acute exacerbation.
Centrilobular emphysema (CLE) defined as permanent enlargement of the airspaces distal to the terminal bronchioles is the most common type of smoking related emphysema,
and is usually upper lung predominant.
CLE results from the dilation and destruction of respiratory bronchioles within a single acinus and coalescence of several primary lesions.
It is characterized by small well-defined or poorly defined areas of low attenuation surrounded by normal lung.
Fleischner Society published a guideline for the scoring of CLE as trace involving less than 0.5% of a lung zone; mild between 0.5% and 5%; and moderate if more than 5% (Fig.
2).
Severe emphysema should be categorized as either confluent (larger areas of low attenuation) or advanced destructive emphysema (ADE) if expansion of the entire secondary lobule,
distortion of the pulmonary architecture or splaying or decreased caliber of vessels is present (Fig.
3).
Panlobular emphysema (PLE) like ADE refers to destruction across the lobule,
however this type of emphysema presents in a younger age group and is most severe in the lower lung zones in two-thirds of the patients (Fig.
4).
It is linked to a genetic defect in the alpha 1-antitrypsin gene that causes the protein to misfold and accumulate in liver cells,
where it stimulates inflammation and subsequent cirrhosis,
without being secreted into the circulating blood.
Paraseptal emphysema (PSE) is secondary to destruction of the distal acinus,
located near the pleural surface,
close to the chest wall and in the interlobar fissures.
Bullae,
defined as a focal lucencies measuring greater than 1 cm in diameter with thin wall,
are common in PLE and can lead to spontaneous pneumothorax.
In addition,
they can become quite large,
leading to severe compression of the remaining lung,
further hampering pulmonary function (Fig.
5).
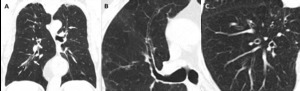
Fig. 1: Bronchial wall thickening due to cigarette smoking in a 79-year-old man. (A) Coronal, (B) and (C) axial images from a chest CT show diffuse thickening of the bronchial walls (arrows). Bronchial wall thickening is an important independent predictor factor of FEV1 and of the risk of acute exacerbation.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
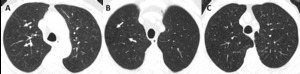
Fig. 2: Fleischner Society guidelines for visually defined subtypes of CLE. Axial images in 3 smokers show (A) trace CLE, with minimal centrilobular lucencies occupying less than 0.5% of a lung zone (arrows). Ground-glass centrilobular nodules are also present in the upper lobes, consistent with RB; (B) mild CLE, defined as scattered centrilobular lucencies (arrows) involving 0.5% to 5% of a lung zone; (C) moderate CLE, with more numerous centrilobular lucencies occupying more than 5% of any lung zone.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
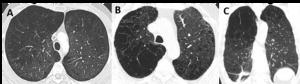
Fig. 3: Fleischner Society guidelines for visually defined subtypes of severe CLE. (A) Axial CT image depicts confluent CLE with coalescent centrilobular lucencies without significant distortion of the pulmonary architecture. (B) Axial and (C) coronal images show ADE, with panlobular lucencies, distortion of the underlying pulmonary architecture and hyperexpansion of the secondary pulmonary lobules.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
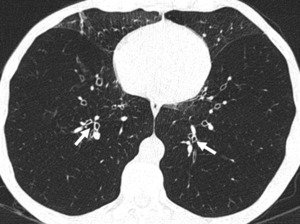
Fig. 4: PLE in a 51-year-old man with alpha-1 antitrypsin deficiency. (A) Axial CT image shows marked hyperinflation and lower lobe predominant emphysema, involving the entire secondary pulmonary lobules. No normal lung architecture is seen in the lower lobes. Bronquial wall thickening and bronquiectasis are also depicted (arrows).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
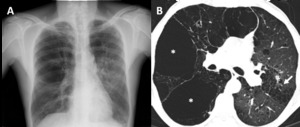
Fig. 5: Bullous emphysema in a 55-year-old man. (A) Posteroanterior (PA) radiograph shows lucencies in the superior half of the right hemitorax and in the upper left hemitorax. (B) Axial CT shows upper lobe predominant bullae (asterisks) along the subpleural surface of the right lung. Moderate CLE is also present (arrow).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
ACUTE EOSINOPHILIC PNEUMONIA
The development of AEP is associated with 3 subpopulations of smokers: new-onset smokers,
smokers who quit and restart smoking,
and also the smokers who increase their daily use of cigarettes.
Patients present with acute febrile illness,
hypoxemia,
and respiratory failure,
requiring intubation.
Bronchoalveolar lavage shows increased numbers of eosinophils.
The damage is caused by the eosinophils at the level of the basement membranes of the capillary endothelium and alveolar epithelium leading to a leakage of fluid,
blood,
and proteinaceous material into the air spaces.
Histopathologic findings show diffuse alveolar damage coupled with interstitial and alveolar infiltrates of eosinophils.
Imaging findings are nonspecific showing lower lung predominant or diffuse consolidation,
ground glass opacity,
scattered nodules,
and septal thickening.
Unilateral or bilateral pleural effusions are present (Fig.
6).
The radiologic differential includes fluid overload,
ARDS,
pulmonary hemorrhage and infection.
Diagnosis is often made by the bronchoalveolar lavage or histopathologic findings.
Patients respond rapidly to the administration of steroids,
usually within days,
with resolution of the immunologic process.
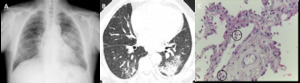
Fig. 6: Radiologic-pathologic correlation of AEP in a 63-year-old man. (A) Posteroanterior (PA) radiograph shows bilateral ground glass opacities and consolidation in the mid right lung zone. A small left pleural effusion is present. (B) Axial CT shows peripheral lower lobe consolidations and ground glass opacities, mainly on the left. (C) Transthoracic lung biopsy showing eosinophils infiltrating the interstitium and the alveolar spaces (circles); oedema and reactive pneumocytes are seen; no necrotizing vasculitis is observed (H&E, x400).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
PULMONARY LANGERHANS CELL HISTIOCYTOSIS
PLCH is an uncommon disease found in approximately 3-4% of smokers.
It primarily affects individuals in the 2nd to 4th decade of life,
and patients most commonly present with dyspnea and cough,
although up to 25% are asymptomatic at the time of disease discovery.
Histopathologic findings reveal discrete bronchiolocentric,
stellate interstitial nodules which in the early stage are cellular,
containing Langerhans cells admixed with variable numbers of lymphocytes,
fibroblasts,
eosinophils,
neutrophils,
plasma cells,
and pigmented macrophages.
Langerhans cells show characteristic deeply grooved nuclei and are immunoreactive with CD1a and S-100.
Over time,
cellular nodules are replaced in a centripetal fashion by fibrous tissue to form cellular and fibrotic nodules and,
in the late stage,
entirely fibrotic bronchiolocentric stellate scars surrounded by distorted and enlarged air spaces.
Imaging reveals upper lobe predominance of nodules and cysts of varying wall thickness and irregular margins,
with relatively sparing of the lung bases and the tips of the middle lobe and the lingula.
Imagiological findings have a typical progression over time from a nodular pattern to a predominance of cysts with bizarre outlines (Fig.
7).
The later stage of the disease is associated with significant emphysematous areas either related to the stellate scaring of PLCH and/or typical smoking-related emphysema (Fig.
8).
Ground-glass attenuation in patients with PLCH correlates with the presence of RB-DIP-like changes.
A critical component in the management of PLCH is smoking cessation.
Smoking cessation often leads to stabilization of symptoms and radiologic abnormalities (Fig.
9).
However,
some individuals may show disease progression leading to respiratory failure despite smoking cessation (Fig.
10).
PLCH should be differentiated from Lymphangioleiomyomatosis (LAM),
in which cysts tend to have a more conventional rounded shape,
with a relatively uniform distribution with no zonal predilection.
Other differentials should also be considered,
such as pulmonary metastasis,
sarcoidosis,
Birt-Hogg-Dubé syndrome,
and infections (Fig.
11).
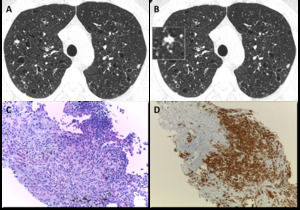
Fig. 7: Pulmonary Langerhans cell histiocytosis in a 40-year-old man. (A) Axial CT image shows upper lobe predominance of nodules and cysts of varying wall thickness and irregular margins. (B) Axial CT image depicting an amplified cellular nodule. (C) Transthoracic lung biopsy of the nodule in B shows a nodular aggregate of cells (lymphocytes, eosinophils, plasma cells and Langerhans cells), centered in bronchioles and extending to the interstitium (H&E, x200). (D) CD1a immunostaining highlights the Langerhans cells (CD1a, x100).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
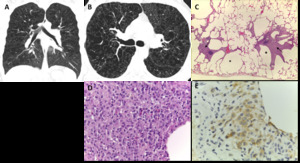
Fig. 8: End-stage PLCH. (A) Coronal and (B) axial CT images in a 30-year-old man show diffuse upper lobe predominant lucencies with some relative basilar sparing. Imagiological findings were thought to represent severe confluent emphysema. (C) Small cysts (*) with nodular aggregates of Langerhans cells in their walls, with a stellate appearance and centered in bronchioles (arrows) (H&E, x40); (D) The cells in the nodules have vesicular nuclei and a pale cytoplasm and sometimes with a reniform appearance; some eosinophils are present (H&E, x400). (E) CD1a immunostaining highlights the Langerhans cells.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
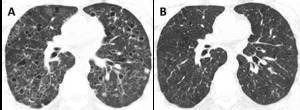
Fig. 9: PLCH. Regression of the imagiological findings over time (2 years interval), after smoking cessation.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
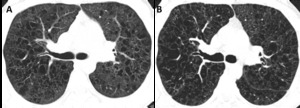
Fig. 10: PLCH. Progression of the imagiological findings over time (10 years interval), despite smoking cessation, in a 30-year-old man.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
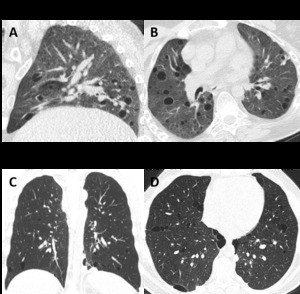
Fig. 11: Differential diagnosis of PLCH. (A) Sagittal and (B) axial CT images show a 50-year-old woman with LAM. There are multiple rounded shaped cysts, with a relatively uniform distribution with no zonal predilection and affecting the lung bases. (C) Coronal and (D) axial CT images depict multiple cysts with a lower zone distribution, in a 35-year-old male with Birt-Hogg-Dubé syndrome. Patient had a history of recurrent pneumothoraces.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
RESPIRATORY BRONCHIOLITIS AND DESQUAMATIVE INTERSTITIAL PNEUMONIA
DIP and RB-ILD are relatively uncommon forms of ILD that are strongly associated with cigarette smoking.
Patients generally present with an insidious onset of dyspnea and cough over a course of weeks or months.
Inspiratory crackles are present in about one half of patients and digital clubbing is present in one fourth of those with DIP as well as of those with RB-ILD.
Most patients with DIP or RB-ILD have a relatively stable clinical course,
but deaths can occur in those with DIP.
RB is a histopathologic lesion found in the lungs of virtually all cigarette smokers.
It is usually asymptomatic and of little clinical significance and differentiated from RB-ILD only by the presence of pulmonary symptoms and abnormal pulmonary function test results.
Histopathologic examination of both entities reveals the presence of typical pigmented (smokers) macrophages centered on the respiratory bronchiole and adjacent alveoli.
DIP reveals smokers macrophages diffusely filling the alveoli,
with associated thickening of the alveolar septa due to inflammatory cells.
Interstitial fibrosis in DIP is usually mild but generally more severe than in respiratory bronchiolitis.
Both entities represent a continuum of the same process,
namely the overproduction and migration of macrophages in the distal bronchioles and alveoli due to smoking.
On imaging,
RB-ILD appears as poorly defined low attenuation centrilobular nodules,
ground-glass opacities,
thickening of the bronchial walls,
few thickened interlobular septa,
and lobular air-trapping on expiratory CT,
with upper lobe predominance of the findings.
Upper lobe emphysema is also commonly present (Fig.
12).
DIP often shows lower lobe predominance of ground glass and reticular opacities interspersed with more normal lung areas,
often creating a mosaic attenuation.
Small cystic spaces may develop within the areas of ground glass opacity.
The distribution of findings is more often subpleural,
but may also be random or diffuse (Fig.13).
Emphysema and findings of RB can be seen.
In DIP,
coexistent areas of pulmonary fibrosis may be present and this finding is associated with disease progression in some individuals,
despite smoking cessation and steroids.
The differential diagnosis includes hypersensitivity pneumonitis,
NSIP,
and atypical infections such as Pneumocystis jiroveci pneumonia (Fig.
14 and Fig.
15).
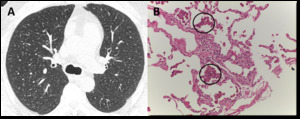
Fig. 12: RB-ILD in a 52-year-old man with a long smoking history. (A) Axial CT image depicts diffuse, ill-defined centrilobular nodules. Due to worsening symptoms, the patient underwent lung biopsy. (B) Excess numbers of alveolar macrophages (circles) with light brown granules filling the cytoplasm, in a predominant centriacinar location; mild interstitial lymphocytic infiltrate (H&E, x200).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
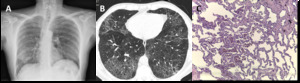
Fig. 13: DIP in a 58-year-old heavy-smoker man. (A) Posteroanterior (PA) radiograph shows reticular opacities and ground glass attenuation in both lungs, predominantly in the lower lung zones. (B) Axial CT image shows ground glass opacities with cystic changes in the lower and mid lobes and lingula. There are well-defined areas of sparing, creating a mosaic attenuation. Mild areas of reticulation and bronchiectasis are also present, signifying fibrosis. (C) Large numbers of alveolar macrophages with light brown granules filling the cytoplasm; mild interstitial lymphocytic infiltrate and mild fibrosis (H&E, x200).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
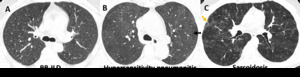
Fig. 14: Differential diagnosis of RB-ILD. (A) Axial CT image shows RB-ILD. (B) Axial CT image depicts subacute hypersensitivity pneumonitis (HP) in a 71-year-old woman with long history of exposition to moldy hay (farmer´s lung). Imagiological findings of HP are similar to RB-ILD, which also appears as centrilobular nodules predominantly in the upper lobes (arrow) and patchy areas of ground-glass attenuation. A clinical history is crucial to make the differential diagnosis. (C) Sarcoidosis in a 37-year-old man. Axial CT image shows diffuse areas of nodularity with peribronchial distribution and also in relation to the subpleural region (orange arrow). There are also some surrounding ground glass opacities.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
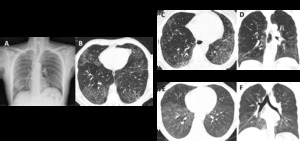
Fig. 15: Differential diagnosis of DIP. (A) and (B) images show DIP. (C) Axial and (D) coronal CT images depict NSIP in a female patient with systemic sclerosis. CT findings show bilateral and asymmetrical ground glass opacities, with a lower lobe predominance and immediate subpleural sparing. (E) Axial and (F) coronal CT images depict pneumocystosis in an immunocompromised patient with HIV. Imagiological findings show bilateral ground glass opacities, predominantly in the lower lobes.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
SMOKING-RELATED INTERSTITIAL FIBROSIS
Cigarette smoke leads to alveolar wall fibrosis or AEF (air space enlargement with fibrosis),
which increases with time and intensity of exposure.
Smokers who have emphysema and fibrosis can be extremely dyspneic out of proportion to their spirometric flow rates,
due to the combined restrictive fibrosis and obstructive physiology seen with small airways disease.
The key to diagnosis is both the physiologic reduction in diffusing capacity and CT findings.
The progression of disease in smokers with this type of fibrosis appears to be slower than in patients with idiopathic pulmonary fibrosis (IPF).
Smoking can cause various degrees of pulmonary fibrosis ranging from patchy alveolar wall fibrosis to diffuse alveolar wall fibrosis.
AEF combines features of emphysema and lung fibrosis.
The characteristic and radiological feature of AEF is subpleural sparing multiple thin-walled cysts (MTWCs),
reticular opacities and ground-glass attenuation.
Radiological differentiation of honeycombing in IPF and MTWCs in AEF may be confusing.
MTWCs in AEF appear as thin walled cysts (less than 1 mm) predominantly located in the upper lobe and upper and middle portion of the lower lobe,
slightly apart from pleura and involving deeper lung tissue.
The progression of fibrosis translates on imaging as increasing lower-lobe predominant ground glass opacities,
reticulation,
and traction bronchiectasis,
which usually represent NSIP (Fig.
16).
These changes are also interspersed with other findings of smoking related injury,
which can include emphysema,
RB,
and DIP.
Foci of CLE may be better defined due to the alveolar wall fibrosis.
The syndrome of combined pulmonary fibrosis and emphysema (CPFE) is a clinically severe pulmonary disease incorporating features of pulmonary emphysema and fibrosis.The histological types of pulmonary fibrosis found in CPFE syndrome are typically described as usual interstitial pneumonia (UIP) or nonspecific interstitial pneumonia (NSIP),
with the latter evident in only a minority of reported cases.
The estimated prevalence of emphysema is thought to range between one quarter and one half of all IPF patients.
CPFE syndrome is most frequently found in older heavy-smoker males (mean age of 65–70 years) and portends a higher risk of development of pulmonary hypertension (between 50% and 90%),
with consequent higher mortality and lower median survival time (25 months) as compared to IPF alone (34 months).
Imagiological findings are centrilobular and/or paraseptal emphysema associated with pulmonary fibrosis in the lower lobes (Fig.
17).
Patients with CPFE had significantly less extensive pulmonary fibrosis than IPF patients without emphysema.
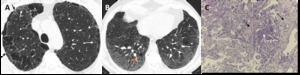
Fig. 16: DIP combined with fibrotic NSIP pattern. (A) Axial CT image through the upper lobes shows CLE and PLE, associated with reticulation (black arrows) and discrete patchy ground glass opacity (white arrows).The areas of emphysema with the areas of ground glass opacity appear to have more well-defined walls, a finding termed as AEF. (B) Axial CT through the lower lobes shows ground glass opacity with scattered cystic changes, mild reticulation and bronchiectasis (orange arrow), signifying underlying fibrosis. (C) Lung transbronchial criobiopsy with uniform thickening of alveolar septa by collagen deposition (black arrows), mild associated inflammation, emphysema and respiratory bronchiolitis (white arrow) (H&E, x100).
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT
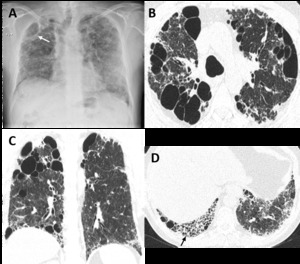
Fig. 17: Combined pulmonary fibrosis and emphysema (CPFE). (A) Posteroanterior (PA) radiograph shows right volume loss, diffuse bilateral reticular opacities and ground-glass attenuation. There are lucencies in the upper lobes (white arrow), signifying bullous emphysema. (B) Axial and (C) coronal CT images show centrilobular, paraseptal, and bullous emphysema predominantly in the upper lobes. (D) Axial image through the lower lobes shows peripheral predominant pulmonary fibrosis with reticulation, traction bronchiectasis and honeycombing (black arrow), in a UIP pattern.
References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT


 Coronal, (B) and (C) axial images from a chest CT show diffuse thickening of the bronchial walls (arrows). Bronchial wall thickening is an important independent predictor factor of FEV1 and of the risk of acute exacerbation. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 trace CLE, with minimal centrilobular lucencies occupying less than 0.5% of a lung zone (arrows). Ground-glass centrilobular nodules are also present in the upper lobes, consistent with RB; (B) mild CLE, defined as scattered centrilobular lucencies (arrows) involving 0.5% to 5% of a lung zone; (C) moderate CLE, with more numerous centrilobular lucencies occupying more than 5% of any lung zone. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image depicts confluent CLE with coalescent centrilobular lucencies without significant distortion of the pulmonary architecture. (B) Axial and (C) coronal images show ADE, with panlobular lucencies, distortion of the underlying pulmonary architecture and hyperexpansion of the secondary pulmonary lobules. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image shows marked hyperinflation and lower lobe predominant emphysema, involving the entire secondary pulmonary lobules. No normal lung architecture is seen in the lower lobes. Bronquial wall thickening and bronquiectasis are also depicted (arrows). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Posteroanterior (PA) radiograph shows lucencies in the superior half of the right hemitorax and in the upper left hemitorax. (B) Axial CT shows upper lobe predominant bullae (asterisks) along the subpleural surface of the right lung. Moderate CLE is also present (arrow). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Posteroanterior (PA) radiograph shows bilateral ground glass opacities and consolidation in the mid right lung zone. A small left pleural effusion is present. (B) Axial CT shows peripheral lower lobe consolidations and ground glass opacities, mainly on the left. (C) Transthoracic lung biopsy showing eosinophils infiltrating the interstitium and the alveolar spaces (circles); oedema and reactive pneumocytes are seen; no necrotizing vasculitis is observed (H&E, x400). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image shows upper lobe predominance of nodules and cysts of varying wall thickness and irregular margins. (B) Axial CT image depicting an amplified cellular nodule. (C) Transthoracic lung biopsy of the nodule in B shows a nodular aggregate of cells (lymphocytes, eosinophils, plasma cells and Langerhans cells), centered in bronchioles and extending to the interstitium (H&E, x200). (D) CD1a immunostaining highlights the Langerhans cells (CD1a, x100). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Coronal and (B) axial CT images in a 30-year-old man show diffuse upper lobe predominant lucencies with some relative basilar sparing. Imagiological findings were thought to represent severe confluent emphysema. (C) Small cysts (*) with nodular aggregates of Langerhans cells in their walls, with a stellate appearance and centered in bronchioles (arrows) (H&E, x40); (D) The cells in the nodules have vesicular nuclei and a pale cytoplasm and sometimes with a reniform appearance; some eosinophils are present (H&E, x400). (E) CD1a immunostaining highlights the Langerhans cells. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
, after smoking cessation. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
, despite smoking cessation, in a 30-year-old man. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Sagittal and (B) axial CT images show a 50-year-old woman with LAM. There are multiple rounded shaped cysts, with a relatively uniform distribution with no zonal predilection and affecting the lung bases. (C) Coronal and (D) axial CT images depict multiple cysts with a lower zone distribution, in a 35-year-old male with Birt-Hogg-Dubé syndrome. Patient had a history of recurrent pneumothoraces. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image depicts diffuse, ill-defined centrilobular nodules. Due to worsening symptoms, the patient underwent lung biopsy. (B) Excess numbers of alveolar macrophages (circles) with light brown granules filling the cytoplasm, in a predominant centriacinar location; mild interstitial lymphocytic infiltrate (H&E, x200). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Posteroanterior (PA) radiograph shows reticular opacities and ground glass attenuation in both lungs, predominantly in the lower lung zones. (B) Axial CT image shows ground glass opacities with cystic changes in the lower and mid lobes and lingula. There are well-defined areas of sparing, creating a mosaic attenuation. Mild areas of reticulation and bronchiectasis are also present, signifying fibrosis. (C) Large numbers of alveolar macrophages with light brown granules filling the cytoplasm; mild interstitial lymphocytic infiltrate and mild fibrosis (H&E, x200). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image shows RB-ILD. (B) Axial CT image depicts subacute hypersensitivity pneumonitis (HP) in a 71-year-old woman with long history of exposition to moldy hay (farmer´s lung). Imagiological findings of HP are similar to RB-ILD, which also appears as centrilobular nodules predominantly in the upper lobes (arrow) and patchy areas of ground-glass attenuation. A clinical history is crucial to make the differential diagnosis. (C) Sarcoidosis in a 37-year-old man. Axial CT image shows diffuse areas of nodularity with peribronchial distribution and also in relation to the subpleural region (orange arrow). There are also some surrounding ground glass opacities. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 and (B) images show DIP. (C) Axial and (D) coronal CT images depict NSIP in a female patient with systemic sclerosis. CT findings show bilateral and asymmetrical ground glass opacities, with a lower lobe predominance and immediate subpleural sparing. (E) Axial and (F) coronal CT images depict pneumocystosis in an immunocompromised patient with HIV. Imagiological findings show bilateral ground glass opacities, predominantly in the lower lobes. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
 Axial CT image through the upper lobes shows CLE and PLE, associated with reticulation (black arrows) and discrete patchy ground glass opacity (white arrows).The areas of emphysema with the areas of ground glass opacity appear to have more well-defined walls, a finding termed as AEF. (B) Axial CT through the lower lobes shows ground glass opacity with scattered cystic changes, mild reticulation and bronchiectasis (orange arrow), signifying underlying fibrosis. (C) Lung transbronchial criobiopsy with uniform thickening of alveolar septa by collagen deposition (black arrows), mild associated inflammation, emphysema and respiratory bronchiolitis (white arrow) (H&E, x100). References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")
. (A) Posteroanterior (PA) radiograph shows right volume loss, diffuse bilateral reticular opacities and ground-glass attenuation. There are lucencies in the upper lobes (white arrow), signifying bullous emphysema. (B) Axial and (C) coronal CT images show centrilobular, paraseptal, and bullous emphysema predominantly in the upper lobes. (D) Axial image through the lower lobes shows peripheral predominant pulmonary fibrosis with reticulation, traction bronchiectasis and honeycombing (black arrow), in a UIP pattern. References: Department of Radiology, Centro Hospitalar São João, Faculdade de Medicina da Universidade do Porto – Porto/PT")