ABBREVIATIONS
Fig. 60
INTRODUCTION
Muscle - gross anatomy
Muscle tissue consists of muscle fibers that are capable of contraction along their longitudinal axis.
Skeletal muscle in particular is a highly specialized organ system evolved for locomotion and energy metabolism in multicellular organisms.
The main elements that constitute a muscle tissue are displayed in Fig.
below.
Other fundamental structures relevant to muscle physiology include tendons,
nerves and blood vessels.
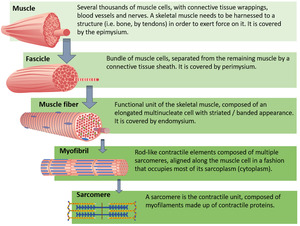
Fig. 2:
Muscle - micro-anatomy
Within each skeletal muscle cell or fiber there are cylindrical structures called myofibrils.
They extend and attach to the cell membrane at either end.
Contraction of the myofibrils result in contraction of the entire cell.
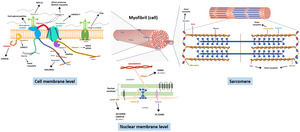
Fig. 3: Molecular structures relevant for understanding hereditary myopathies.
Dystrophin,
DAPC and hereditary myopathies
Myofibrils are made-up of myofilaments,
which are organized in repeating units along the length of the myofibril called sarcomeres.
Further description of a sarcomere and its mechanism of action for allowing muscle contraction is described in Fig.
above (right).
The key structure in muscular dystrophies is the muscle cell membrane (sarcolemma).
Most muscular dystrophies are due to breakdown of the dystrophin-associated glucoprotein complex (DAPC),
a network of fibrous proteins that bind myofibers to the matrix and stabilize the sarcolemma during contraction and relaxation (see Fig.
above,
left).
Dystrophin connects the sarcolemma to the actin cytoskeleton,
playing an important role in muscle cell membrane stability.
The connection is achieved through the DAPC.
Loss of integrity of this network causes stress fractures of the sarcolemma to develop during muscle contraction.
Influx of calcium through these breaks leads to myonecrosis.
Atrophied muscle is further substituted by fibrous and fatty tissues,
one of the hallmarks of muscular dystrophy.
Also,
disruption of sarcolemma causes leakage of muscle proteins such as creatine kinase into the serum.
Other hereditary myopathies are associated with disruption of proteins present in the nuclear envelope (see Fig.
above,
down).
Imbalance between muscle degeneration and regeneration of skeletal muscle leads to inflammatory reponses and bursts of cytokine expression,
which contribute to fibrosis and fatty deposition observed as disease progresses.
RELEVANT SECTIONAL ANATOMY
In this section,
we present some of the most relevant muscle anatomy one needs to be acquainted with when assessing a WBMRI.
Thigh sectional anatomy
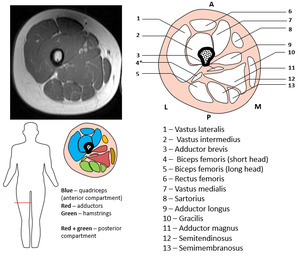
Fig. 4: Sectional anatomy - mid-portion of the thigh
Lower leg/calf sectional anatomy
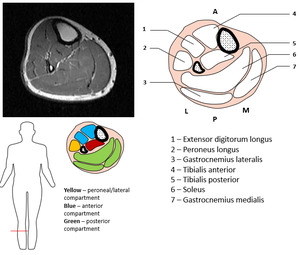
Fig. 5: Sectional anatomy - mid-portion of the lower leg.
Whole body MRI in 7 muscles or less
The following gif shows some of the most relevant muscles that should be addressed at different levels of the human body in a whole body MRI (focusing on 7 or less muscles per level so not to overburden the reporter).
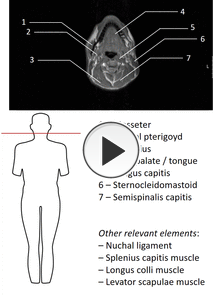
Fig. 6: Relevant muscle anatomy in WBMRI
PATHOLOGIC FINDINGS
Major muscular manifestations of the diseases described in this presentation are defined below.
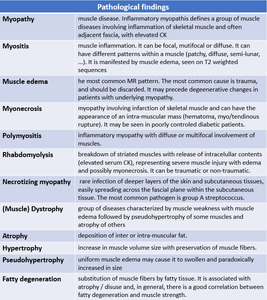
Fig. 58

Fig. 59
CLASSIFICATION OF DISEASES
This presentation approaches several neuromuscular disorders,
which are described on the table below.
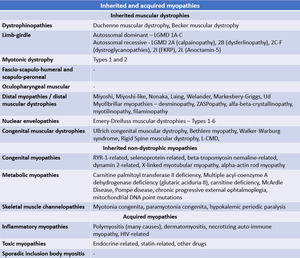
Fig. 53
Muscular dystrophies
Muscular dystrophies are genetically transmitted diseases characterised by degeneration and loss of myofibers and,
clinically,
by inexorably progressive weakness and,
many of them,
by elevated CK.
The pattern of weakness,
tempo of evolution,
and mode of inheritance vary among different dystrophies.
The most common muscular dystrophies include Duchenne muscular dystrophy (DMD),
Becker muscular dystrophy (BMD),
facioscapulohumeral muscular dystrophy (FSHD),
and myotonic dystrophy.
In adults,
the most common dystrophies are myotonic dystrophy and the limb girdle dystrophies.
Duchenne muscular dystrophy is the most common form of muscular dystrophy in childhood,
characterized by absent production of dystrophin,
a protein important for muscle fiber strength by linking actin filaments to structrual proteins.
In Becker muscular dystrophy,
the reading frame of the dystrophin gene is preserved and thus there is a milder clinical phenotype,
as there is some (insufficient) production.

Fig. 63
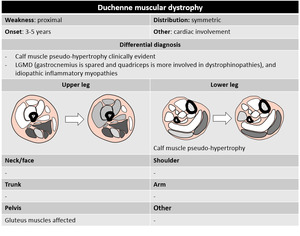
Fig. 7
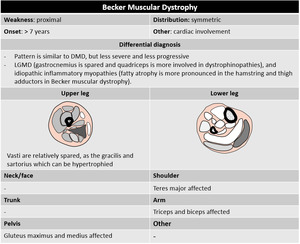
Fig. 8
- Limb-girdle muscle dystrophies
Genetically heterogeneous group of conditions associated with recessive and dominant inheritance,
characterized clinically by proximal weakness often pronounced in the hip girdle,
sparing of facial and extraocular muscles,
and variable cardiorespiratory involvement.
Prevalence of specific LGMDs is unknown in Portugal,
but our neighbourest country,
Spain,
shows predominance of the LGMD 2A form.

Fig. 64
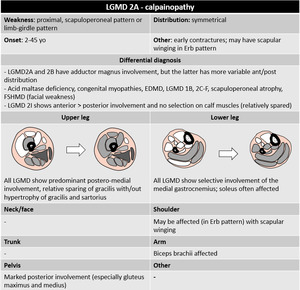
Fig. 38

Fig. 39
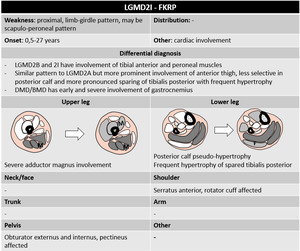
Fig. 40
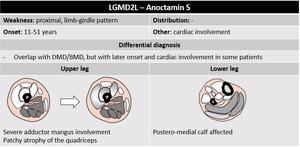
Fig. 41
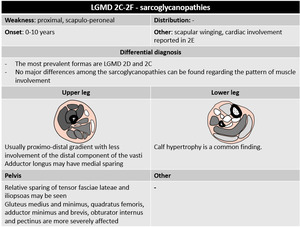
Fig. 42
Dominantly-inherited LGMDs (1A-C) show considerable overlap with other disorders:
- LGMD1A – myofibrillar myopathy
- LGMD1B – emerinopathies
- LGMD1C – rippling muscle disease,
asymptomatic hyperCKemia and a distal myopathy.
Biopsy – non-specific dystrophic and inflammatory changes that cause diagnostic confusion; histochemical studies may be helpful.
Diagnosis – muscle imaging remains cumbersome for differential diagnosis within this group of disorders.
MRI is useful to distinguish late-onset acid maltase deficiency,
occasionally presenting as a phenocopy of recessive LGMDs but showing unusually severe and early adductor involvement
- Facio-scapulo-humeral dystrophy
FSHD is the 3rd most common muscular dystrophy and the most prevalent muscular dystrophy (7:100,000).
Onset of symptoms start during adulthood and the condition is characterized by asymmetric loss of strength and atrophy of muscular tissue starting in the face and shoulder region.
Distribution,
however,
varies significantly among patients.

Fig. 65
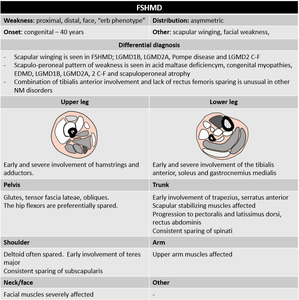
Fig. 43
- Oculopharyngeal muscular dystrophy
Oculopharyngeal muscular dystrophy is a late-onset myopathy accompanied by ptosis and dysphagia.
Patients can eventually develop proximal muscle weakness.

Fig. 66
The group of emerinopathies/laminopathies comprise multiple muscular dystrophies associated with mutations of nuclear envelope components,
that typically present with weakness in a scapuloperoneal or limb-girdle distribution,
contractures and defects in cardiac conduction and/or cadiomyopathy.

Fig. 67
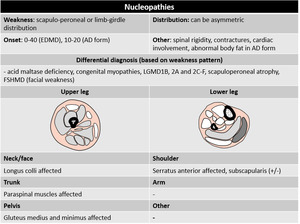
Fig. 44
- Myotonic dystrophy / dystrophic myotony
Myotonic dystrophy type 1 and type 2 are autosomal dominant myopathies.
DM1 is the most common adult muscular dystrophy.
Patients present with muscle weakness and stiffness,
variable degrees of cognitive impairment,
endocrine abnormalities,
cataracts,
cardiac conduction abnormalities and/or cardiomyopathy.
DM2 is a milder form that shares several features with DM1.

Fig. 68
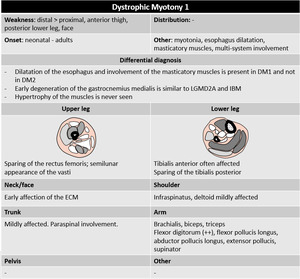
Fig. 45
Pattern DM2 – similar to DM1,
with less severe fatty degeneration.
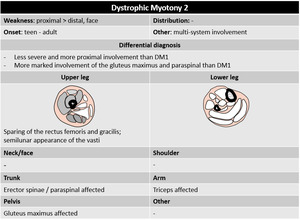
Fig. 46
Distal weakness is an uncommon phenomenon in myopathies,
so when it is present there is a “limited” differential diagnosis that fits in the diseases in this category.
Distal myopathies may be separated in terms of age of onset (early-onset and late-onset).
We will not go into further detail regarding most of the distal myopathies,
except for the group of myofibrillar myopathies,
displayed below.
Myofibrillar myopathies
These conditions are grouped together due to their similarities on histological examination,
with degradation of myofibrils and accumulation of degradation products in intracellular inclusions.
They are late-onset disorders with often prominent distal weakness and variable multisystem involvement (especially cardiac conduction abnormalities).

Fig. 69

Fig. 61
- Congenital muscle dystrophies
The congenital muscular dystrophies (CMDs) are a clinically and genetically heterogeneous group of disorders that present at birth or within the first few months of life,
with hypotonia,
muscle weakness,
contractures and motor developmental delay.
Unlike congenital myopathies,
they are progressive and often share brain and other organ abnormalities,
as well as dystrophic changes on muscle histology.

Fig. 70
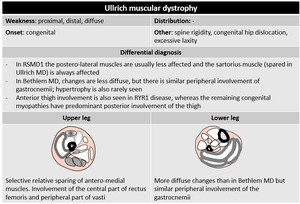
Fig. 49
Inherited non-dystrophic myopathies
Congenital myopathies are genetic muscle disorders characterized by structural abnormalities of myofibers and/or accumulation of abnormal protein in the sarcoplasm.
They are defined by non-progressive weakness and hypotonia presenting in the first year of life.
The most common CMs are core myopathies,
nemaline myopathy and centronuclear myopathy.
Among the core myopathies,
the most frequent genetic change is RYR1 mutation.

Fig. 71
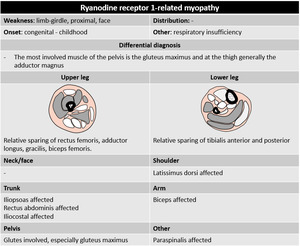
Fig. 47
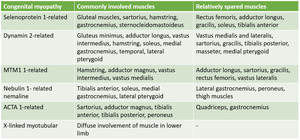
Fig. 48
Metabolic myopathies may be divided into lipid metabolic,
glycogen metabolic and mitochondrial disorders.
Symptoms may be provoked by exercise or any factor that induces metabolic stress.
If multiple organic systems seem involved,
mitochondrial disorders should be considered.
Muscle biopsy - often helpful in showing excessive glycogen (for glycogen disorders),
excessive lipid droplets (for glycogen disorders),
or ragged red fibers,
which represent focal regions of mitochondrial clumping that result from mitochondria proliferation and dysfunction.
Pattern – non-specific (Pompe disease in table below).
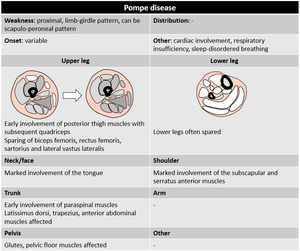
Fig. 50
- Skeletal muscle channelopathies
Skeletal muscle channelopathies manifest clinically with myotonia and may be associated with muscle weakness.

Fig. 72
Acquired myopathies
- Dermatomyositis and polimyositis
Polymyositis is a rare auto-immune and sometimes paraneoplastic inflammatory myositis that most commonly affects patients in their 4th decade of life and women twice as men.
The term “polymyositis” is applied when the condition spares the skin,
and “dermatomyositis“ when polymyositis is associated with a characteristic skin rash.
These conditions typically show insidious diffuse symmetrical proximal involvement of the lower limb girdle and progressing to involve the proximal upper limb girdle,
neck flexor and pharyngeal muscles.
Thus,
muscle edema most often involves the quadriceps and hamstring muscles and,
in some cases,
triceps brachii and deltoid muscles.
In DM,
edema can be focal or diffuse in the muscle,
along the fascia,
subcutaneously or cutaneously.
Fasciitis is common and can be evidenced on US.

Fig. 73
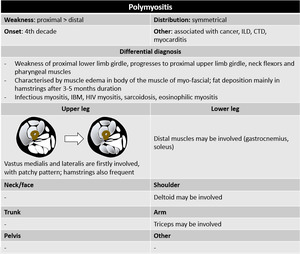
Fig. 52
- Sporadic inclusion body myositis
Sporadic IBM is the most common form of acquired myopathy in the elderly (over 50 years) and is characterized by slowly progressive weakness and atrophy predominantly affecting the quadriceps and deep finger flexor muscles in the forearm,
followed by involvement of proximal muscles and dysphagia.
Inflammation is not a prominent feature and there are no skin changes.
There is often asymmetrical fatty infiltration of skeletal muscles; muscle edema is less prominent.
The legs are more often and more severely affected than the arms.

Fig. 74
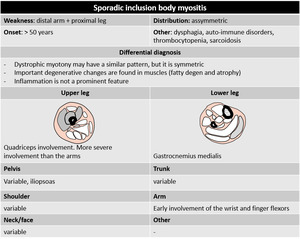
Fig. 51
- Necrotizing auto-immune myopathy
This condition is associated wit anti-SRP or anti-HMGCR antibodies.
Muyscle MRI of the thigh shows muscle edema of both the anterior and posterior compartment with predominant involvement of the vastus lateralis compared to the vastus intermedius.
Fatty infiltration may also be seen in one third of patients,
predominantly of the posterior thigh muscles.
It is more pronounced than in PM.
Assessing on MRI before muscle biopsy yields higher accuracy of the latter,
since the inflammation is often focal.
CK levels are usually highly elevated.
Several substances can cause a myopathic response with inflammatory properties,
most frequently drugs such as steroids,
statins and alcohol.
Imaging shows non-specific muscle edema of the thigh reflecting either rhabdomyolisis or other chronic changes.
Differentiation is difficult with MRI alone.
SUMMARY OF HEREDITARY AND ACQUIRED MYOPATHIES
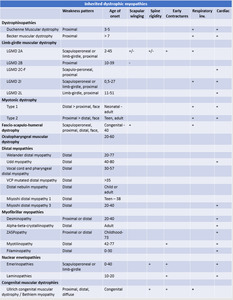
Fig. 54
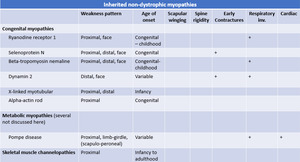
Fig. 55





