In 2013,
the revised American Thoracic Society/European Respiratory Society classification of idiopathic interstitial pneumonias distinguished two rare IIPs: Idiopathic lymphoid interstitial pneumonia (LIP) and idiopathic pleuroparenchymal fibroelastosis (PPFE).
[1]

Fig. 1: Revised ATS/ERS classification of idiopathic interstitial pneumonias.
References: Department of Radiology, Centro Hospitalar de São João - Porto/PT
As these conditions are still poorly recognized,
they pose a diagnostic challenge so,
these societies also highlighted the importance of a multidisciplinary approach to their correct diagnosis that encompasses clinical,
radiologic and pathologic data.
In the work-up of IIPs,
only a multidisciplinary team will confidently decide which patients need a diagnostic biopsy and subsequently,
integrate the pathological findings to the adequate clinical/imagiological context.
|
PLEUROPARENCHYMAL FIBROELASTOSIS
|
PPFE is characterised by elastotic fibrosis of the pleura and subpleural lung parenchyma,
with an upper lobe predilection (typically apical).
Even though it was described an association with lung/bone marrow transplantation,
recurrent infections,
chemotherapeutic drugs,
autoimmune diseases and genetic factors,
most cases are considered idiopathic.
It is an adult pathology,
with a median age of incidence of 53 years [4] and with no gender predilection.
Most patients have no smoking history.
PPFE may have a long subclinical stage without symptoms,
although it usually progresses to exertional dyspnea and dry cough,
and frequently results in pneumothorax. As with other fibrotic lung diseases,
PPFE most commonly presents a restrictive ventilatory pattern and a reduced diffusing capacity of carbon monoxide.
Despite treatment,
disease progression occurs in 60% of the cases,
resulting in death in approximately 40%.[1,2]
|
LYMPHOID INTERSTITIAL PNEUMONIA
|
LIP is a rare benign/reactive lymphoproliferative disease confined to the lungs,
characterised by a dense lymphoid interstitial infiltrate with substantial alveolar wall involvement.
Its pathophysiology is still poorly understood and,
unlike PPFE,
LIP is uncommonly idiopathic,
being most often linked with other underlying disorders such as: autoimmune (specially Sjögren’s syndrome),
infection,
immunodeficiency (HIV),
bone marrow transplantation and drug toxicity.
The median age of incidence is around 47 years [12],
with female gender predominance.
75% of patients have no smoking history [12].
Respiratory symptoms,
such as dyspnea and dry cough,
are more pronounced than constitutional ones.
These patients usually demonstrate a restrictive ventilatory pattern with reduced diffusion capacity and T-cell lymphocytosis in the bronchoalveolar lavage.
Little is still known about this condition’s management and prognosis.
However,
S-I.
Cha et al,
suggested that LIP may have an independent impact on survival,
with a median survival of 11,5 years in their cohort.
The risk of malignant transformation to lymphoma is presumably low.
A summary of the overview of these diseases is found in Fig.
2.
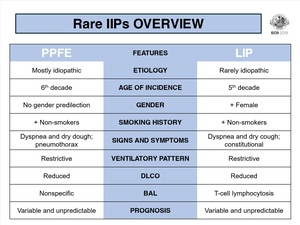
Fig. 2: Rare idiopathic interstitial pneumonias overview.
References: Department of Radiology, Centro Hospitalar de São João - Porto/PT
