REVISIÓN DEL TEMA
EPIDEMIOLOGIA [i](fig 2)
[i](fig 2)
Incidencia aproximada de homocigotos (casos) 1:42000 de la población total (1:20000 varones) e incidencia estimando tanto homocigotos como heterocigotos (portadores) aproximadamente 1:17000.

Fig. 2
PATOGENIA[ii]
Trastorno hereditario del metabolismo de los peroxisomas.
El defecto fundamental es una alteración funcional de los peroxisomas que provoca betaoxidación defectuosa de los ácidos grasos saturados de cadena muy larga (AGML) que así se acumulan en sangre y tejidos.
El proceso dismielinizante es un proceso microglial mielinolítico,
resultante de una vigorosa respuesta inflamatoria al acúmulo de los AGML,
con degeneración axonal(fig 3,
4).
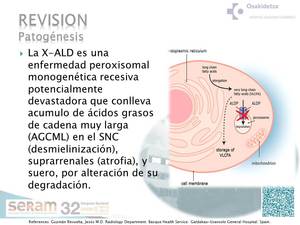
Fig. 3
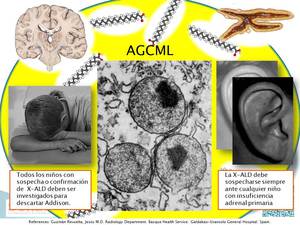
Fig. 4
HERENCIA
Es una enfermedad monogenética con herencia ligada al cromosoma X (como al menos el 50% de las heterocigotas adultas desarrollarán un síndrome AMN-like no es del todo correcto denominarla «recesiva»).
Todas las hijas de hombres afectados son obligadamente heterocigotas.
Cuando es la mujer la portadora existe un 50% de probabilidades en cada embarazo de que el gen defectuoso se transmita a su hijo o la hija.
La clave está en detectar las portadoras heterocigotas y los varones homocigotos pre sintomáticos.No existe correlación genotipo-fenotipo,resultando una gran heterogenidad puesta de manifiesto por la coexistencia de variables fenotipos dentro de una misma familia sin correlacionarse con el gen mutado ni la severidad de las alteraciones bioquímicas.).
La frecuencia de las mutaciones de novo se estima en el 5%(fig 5,
6).
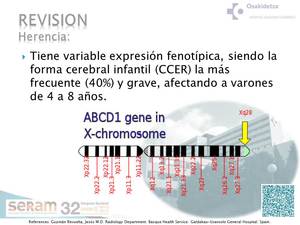
Fig. 5
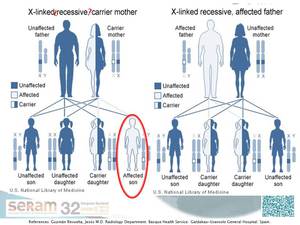
Fig. 6
PRESENTACION CLINICA[iii]
La expresión fenotípica de la enfermedad es variable y no se puede predecir el fenotipo en base a estudios genéticos ni bioquímicos.
Por motivos clínicos se clasifican en 5 fenotipos fundamentales: cerebral infantil,
cerebral adolescente,
adrenomieloneuropatía AMN,
cerebral adulto,
y solo Addison con hiperpigmentación cutaneomucosa(fig 7,
8).La forma clásica cerebral infantil comienza con alteraciones conductuales,
visuales,
auditivas y motoras con progresión rápida que conduce a discapacidad,
tetraparesia espástica,
estado vegetativo y muerte prematura a los 10 años de edad aproximadamente.
Los fenotipos «enfermedad de Addison aislada» tienen riesgo de desarrollar afectación neurológica por lo que se establecerá una vigilancia estrecha.
La información basada exclusivamente en la edad y la puntuación del índice de severidad por RM en el primer contacto es muy predictiva del curso clínico futuro,
y permite seleccionar los candidatos alTrasplante de Células Madre Hematopoyéticas (TCMH) u otras terapias experimentales.

Fig. 7
References: Orphanet Journal of Rare Diseases. 2012; 7:51.
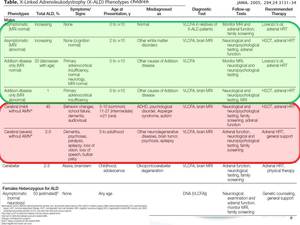
Fig. 8
References: JAMA. 2005, 294;24:3131-34
DIAGNOSTICO DIFERENCIAL[iv]
El patrón de RM cerebral de la X-ALD normalmente es fácilmente reconocido por los neuroradiólogos.Otras enfermedades con similar patrón de RM son las alteraciones en la biogénesis de los peroxisomas,
y el déficit de ACOX1 o DBP con desarrollo tardío de los síntomas y muy distinta presentación clínica por lo que no ofrecen dudas.Los errores retrasos en el diagnostico son más frecuentes cuando la desmielinización ocurre en los lóbulos frontales (los déficits enla planificación,
en el razonamiento ejecutivo,
y en la atención son a menudo los síntomas que conducen al error diagnostico como un TDAH).
Los pacientes con afectación occipital pueden tener clínica similar aunque los síntomas sean menos marcados,
debido a la falta de conexión entre los lóbulos occipito-parietal y frontal(fig9).

Fig. 9
TRATAMIENTO[v],[vi]
En la actualidad se recomienda el tratamiento estándar con TCMH tras el diagnóstico definitivo de la forma cerebral de X-ALD (genético + RM) sin (o con pocos) síntomas neurológicos.
[Grado 1B de evidencia].
En la práctica siguen siendo un desafío por la estrecha ventana terapéutica.
Aproximadamente la mitad de las casos con X-ALD fenotipo cerebral diagnosticados clínicamente están en un estado tan avanzado en el momento del diagnóstico que no son candidatos a tratamientos modificadores de la enfermedad.
En enfermedad pre sintomática (solo elevación de niveles de AGCML y sin clínica ni lesiones RM) es mejor el tratamiento con aceite de Lorenzo (precursores exógenos que al competir por el mismo sistema enzimático para la elongación microsomal de los ácidos grasos saturados de cadena corta a AGCML reduce la carga de AGCML) que la ausencia de tratamiento (Grado 2C de evidencia).Debe de considerarse queel TCMH no corrige la insuficiencia suprarrenal,
a la hora de establecer los tratamientos sintomáticos y/o paliativos correspondientes.
Recientemente se están empleando con éxito terapias genéticas utilizando vectores lentivirales(fig 10).
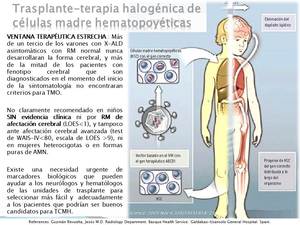
Fig. 10
References: Science. 2009 Nov 6;326(5954):818-23.
[i] Coll MJ,
et al.
X-linked adrenoleukodystrophy in Spain.
Clin Genet 2005;67:418-24.
[ii] Johannes Berger et al.
X-linked adrenoleukodystrophy: Clinical,
biochemical and pathogenetic aspects.Biochimica et Biophysica Acta 1763 (2006) 1721–1732.
[iii] Hugo W.
Moser,
et al.Adrenoleukodystrophy.
New Approaches to a Neurodegenerative Disease.JAMA.
2005,
294;24:3131-34.
[iv] Levers CE et al.Avoiding the Misdiagnosis of Adrenoleukodystrophy: Distinguishing ALD from ADD/ADHDJ Dev Behavior Pediatrics.
1999;20: 31-35.
[v]Aubourg P et al.Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy.
Science.
2009 Nov 6;326(5954):818-23.
[vi] Nathalie Cartier; Patrick Aubourg.
Hematopoietic Stem Cell Transplantation and Hematopoietic Stem Cell Gene Therapy in X-Linked Adrenoleukodystrophy.
Brain Pathology 2010;20:857–862.









:818-23.")