Infections
It is widely demonstrated that IPF patients are more susceptible to pulmonary infections (fig.1)
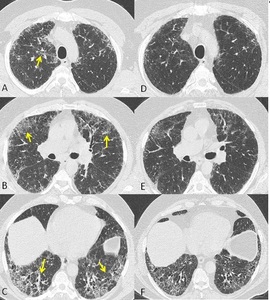
Fig. 1: Axial chest CT scans (A,B,C show patchy areas of ground glass opacities superimposed to a UIP pattern (arrows in A,B,C) , due to infection. Follow up chest CT scans (D,E,F) after two months showing resolution of ground glass opacities.
particularly to Mycobacterium tuberculosis,
Aspergillus species,
and a number of opportunistic infection such as Pneumocystis jirovecii (PJ) and Cytomegalovirus (CMV).
Opportunistic infections manifest specially in patients receiving corticosteroid therapy even if of relatively short duration.
In diffuse fibrotic lung disease the pre-existing fibrotic alteration may modify the typical HRCT presentation of the most common pulmonary infections.
The radiological manifestation of pulmonary tuberculosis (TB) in IPF patients is frequently characterized by mass-like peripheral lesions,
and segmental or lobar consolidation with or without cavitation; centrilobular nodules are rare.
A good knowledge of this atypical pattern of presentation is fundamental because peripheral mass-like lesions can be confused with IPF-associated lung cancer and the TB infection should be immediately treated.
Aspergillous species are ubiquitous pathogenic microorganisms.
Aspergillus infection depends upon host’s immune response,
local lung architecture and concentration of inoculum.
IPF patients are more susceptible to develop two form of Aspergillosis: aspergillomas result from saprofitic colonization of pre-existing lung cavity without tissue invasion and chronic airway invasive aspergillosis well known as necrotizing or semi-invasive aspergillosis.
There is an overlap between this two entities and not always is simple to distinguish this two form.
Aspergillomas on HRCT appears as solid round or oval mass also known as “fungus ball” arising from the cavity wall (fig.2,3);
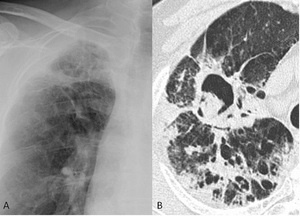
Fig. 2: Micetoma in a patient with biopsy-proven usual interstitial pneumonia. A chest-ray magnification (A) shows a disomogeneous hyperlucency area in the right apical region. Chest CT scan (B) confirms the presence of a micetoma in the right upper lobe.
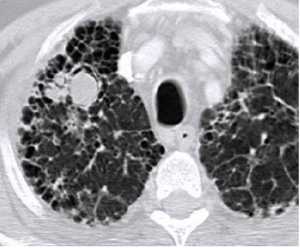
Fig. 3: CT scan showing a mycetoma in the right upper lobe in a 57-year-old man with a combined pulmonary emphysema-fibrosis syndrome.
typically aspergillomas moves when patient change position.
Necrotizing or semi-invasive aspergillosis appears as focal consolidation more frequent localized at lung apices,
with adjacent pleural thickening progresses to cavitation because of local fungus invasion.
Infections caused by PJ pneumonia and viruses infections such as CMV(fig.4) are characterized by diffuse,
symmetric and extensive ground glass opacity (GGO),
consolidation and septal thickening.
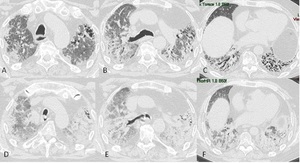
Fig. 4: Images from a 60-year-old man with biopsy-proven usual interstitial pneumonia complicated by Cytomegalovirus pneumonia (CMV). HRCT scans (A,B,C) at baseline. HRCT scans (D,E,F) 2 months later showing diffuse ground-glass opacification superimposed on the fibrotic lung disease.
This alterations in PJ pneumonia are typically distributed in perihilar and upper zone with sparing of subpleural region.
This findings are very similar to acute exacerbation images and frequently it is difficult to distinguish between this two complications; for this reason infection should be confirmed or excluded with endotracheal aspirate or bronchoalveolar lavage (BAL).
Malignances
IPF is also recognized as an independent risk factor for lung cancer: several studies shows that prevalence of lung cancer is five times greater in IPF patients than in the age-matched general patients without IPF.
Pathogenesis of lung cancer in these patients is unclear,
it has been suggested that inflammatory mediators may causes repeated episodes of cellular and genetic damage to respiratory epithelium with occurrence of atypical and dysplastic changes and so development of malignancy.
In IPF patients small cell and squamous cell cancer seems to be relatively more common than adenocarcinoma.
It is also important to remember that synchronous tumors occur more frequently in patients with IPF than in general population.
Tumors on HRCT appears as round or oval solid lesions (fig.5) located at the interface between fibrotic cyst and normal lung or in the midst of subpleural fibrotic cysts of the lower lobes,
less frequently are found in the upper lobe; part solid tumors were seen in adenocarcinoma.
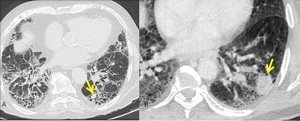
Fig. 5: Chest CT scans (A,B) of two different patient showing the presence of a lung cancer in the left lower lobe (arrows in A,B)
Sometimes tumors may present with indeterminate pattern such as pleural thickening very similar to mesotelioma or GGO within honeycombing that histologically corresponds to mucinuos bronchioloalveolar carcinoma.
It is often difficult to distinguish malignances from areas of confluent fibrosis specially when lesions are located at interface between fibrotic cyst and normal lung or in the midst of subpleural fibrotic cysts,
for this reason is fundamental a meticulous comparison with previous exams.
Patients with IPF only have a poor prognosis,
patients with IPF and cancer have a prognosis even worse because surgical resection,
chemotherapy and radiotherapy could cause acute exacerbation; a regular follow-up and so the early detection of lung cancer are important to give an extra chance to IPF patients(fig.6).
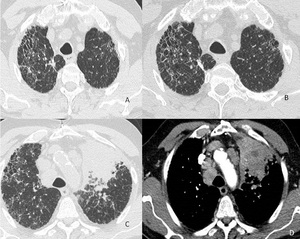
Fig. 6: Images from a 72-year-old man with biopsy-proven usual interstitial pneumonia. HRCT scans (A,B) at baseline. HRCT scans (C,D) 6 years later showing advanced lung cancer in upper left lobe. The patient refused to do CT follow-up at the last 6 years.
Acute exacerbation
Acute exacerbation is a serious complication of IPF that can lead to death in a period ranging from few week to few months.
It may occur at each stage in the natural history of the disease.
Diagnosis of this complication requires previous or current diagnosis of IPF; development of dyspnea or its worsening within 30 days without a recognizable cause,
such as infection,
left heart failure or pulmonary embolism; newly developing bilateral consolidation or ground glass opacity superimposed on a background reticular or honeycombing pattern consisting with UIP.
Acute exacerbation may be idiopathic or may be consequent to thoracic biopsy or resection,
chemotherapy,
radiotherapy and infections.
The newly developing pulmonary opacities can be classified into peripheral,
multifocal,
and diffuse pattern according to the classification of Akira and coworkers.
In peripheral pattern,
parenchymal opacification appeared in the inner peripheral zone adjacent to preexisting subpleural honeycombing or was increased in areas with preexisting peripheral interstitial opacity.
In multifocal pattern,
parenchymal opacification was apparent in central and peripheral regions; however,
the multifocal abnormalities were at a few sites and limited in extent.
Diffuse pattern had generalized pulmonary involvement but regional inhomogeneity(fig.7,8,9).
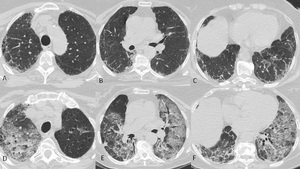
Fig. 7: Acute exacerbation in a 67-year-old man with a UIP pattern. HRCT scans (A,B,C) at baseline. HRCT scans (D,E,F) showing a typical diffuse pattern of acute exacerbation.
References: Courtesy of L.Calandriello, Catholic University, Rome.
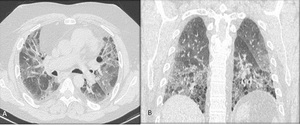
Fig. 8: Acute exacerbation in a 73-year-old man with a UIP pattern. Axial HRCT (A) and coronal MPR reconstruction (B) images showing a diffuse pattern of acute exacerbation.
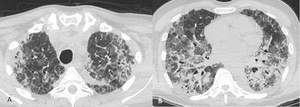
Fig. 9: Acute exacerbation in a 69-year-old man with a UIP pattern. Axial CT (A,B) images showing a multifocal pattern of acute exacerbation
References: Courtesy of L.Calandriello, Catholic University, Rome.
Multifocal and diffuse patterns have been associated with diffuse alveolar damage (DAD),
thus with worse prognosis.
Multifocal pattern is considered to be the early phase of diffuse pattern.
Peripheral pattern does not evolve into diffuse pattern for a short period.
In the acute exacerbation of IPF,
two basic HRCT patterns may be classified: new areas of parenchymal opacification mainly within the peripheral region with relatively limited damage,
and new areas of parenchymal opacification that spread rapidly throughout the lung with a fulminant clinical course.
Diffuse pattern corresponded to DAD at pathology,
whereas peripheral pattern mainly correlated with OP or numerous fibroblastic foci.
The HRCT extent and HRCT patterns has a higher predictive value regarding patient survival than clinical and laboratory data.
HRCT patterns are useful for predicting patient prognosis in acute exacerbation of IPF.
Pulmonary Hypertension
Pulmonary hypertension (PH) is a progressive disease of pulmonary arteries defined as mean pulmonary arterial pressure >25 mmHg at catheterization of the right side of the heart.
It has been reported that prevalence of PH is 20–46% in IPF patients who undergoing work-up for lung transplantation although the prevalence in the wider IPF population is not well documented.
This condition impact adversely on prognosis in IPF with a two to three fold increase on mortality.
The main factors responsible for HP in IPF patients are hypoxia,
noted to be the most efficient vasoconstrictor,
and destruction of lung vasculature that cause vascular remodeling.
Cardiac magnetic resonance (MR) and electrocardiographically (ECG) gated multidetector computed thomographic pulmonary angiography (CTPA) are the best image technique to evaluate anatomical changes that occur in patients with PH, but also on HRCT there are some findings suggestive of pulmonary hypertension.
On transverse images the main pulmonary artery with a diameter of 29 mm or more evaluated at the level of its bifurcation orthogonal to the long axis in mediastinum window setting is suggestive for PH,
but it should not be forgotten that in patients with pulmonary fibrosis main pulmonary artery dilatation may occur in the absence of PH and then the main pulmonary artery diameter at HRCT is an unreliable indicator of PH.
Increased ratio of pulmonary artery diameter to ascending aorta diameter of 1 (fig.10) and a segmental artery-to-bronchus diameter ratio of 1:1 or more in three or four lobes are a more sensible indices of PH.
Furthermore,
demonstration of the main pulmonary artery at the level of the aortic arch is specific to severe PAH (the“egg and banana” sign).
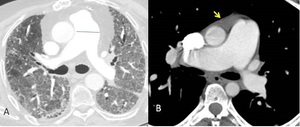
Fig. 10: Axial contrast-enhanced CT scans show an enlargement of main pulmonary artery (diameter3.6 cm) (A) and a pulmonary artery/aorta ratio>1. A minimal pericardial effusion is also seen (arrow in B).
Despite this findings,
it is important to underline that early diagnosis of PH is not a domain of HRCT.
Spontaneous pneumothorax and pneumomediastinum
IPF patients often develop spontaneous pneumothorax and pneumomediastinum that are poorly tolerated.
HRCT is the best technique to diagnose this two entities and to evaluated extension and distribution.
Spontaneous pneumothorax and pneumomediastinum frequently worsen the disease’s course of IPF (fig.11,12).
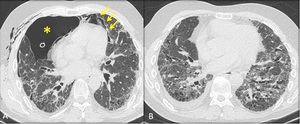
Fig. 11: Axial HRCT scan (A) at the level of the left atrium revealed a right pneumothorax (asterisk) and pneumomediastinum (yellow arrows). Follow-up chest HRCT scan (B) revealed resolution of the pneumothorax. The patient is candidate to lung transplantation.
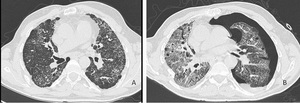
Fig. 12: Images from a 55-year-old man with biopsy-proven usual interstitial pneumonia. HRCT scan (A) at baseline and axial HRCT image (B) after two months demonstrates spontaneous left pneumothorax and acute exacerbation. The patient died.
Disseminated dendriform pulmonary ossification
Disseminated dendriform pulmonary ossification (DPO) are a form of metaplastic mature bone formation in the context of chronic fibrotic alteration of IPF.
DPO is rare finding without clinical manifestations.
DPO are seen on HRCT as multiple tiny calcifications (fig.13) beyond anatomical or lobular configuration that correspond histopathologically to multiple dendriform nodules of mature bone fixed in fibrous stroma in basal,
subpleural areas of fibrosis and honeycombing.
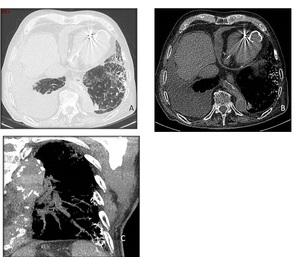
Fig. 13: Dendriform ossification in a 78-year-old man with fibrotic lung disease of unknown cause. Axial CT images with lung (A) and bone window setting (B) show numerous small punctate and branching dense opacities, mainly in fibrotic lung. Oblique MIP projection (C) demonstrates the branching nature of the heterotopic bone formation within areas of fibrosis.

 , due to infection. Follow up chest CT scans (D,E,F) after two months showing resolution of ground glass opacities.")
 shows a disomogeneous hyperlucency area in the right apical region. Chest CT scan (B) confirms the presence of a micetoma in the right upper lobe.")

. HRCT scans (A,B,C) at baseline. HRCT scans (D,E,F) 2 months later showing diffuse ground-glass opacification superimposed on the fibrotic lung disease.")
 of two different patient showing the presence of a lung cancer in the left lower lobe (arrows in A,B)")
 at baseline. HRCT scans (C,D) 6 years later showing advanced lung cancer in upper left lobe. The patient refused to do CT follow-up at the last 6 years.")
 at baseline. HRCT scans (D,E,F) showing a typical diffuse pattern of acute exacerbation. References: Courtesy of L.Calandriello, Catholic University, Rome.")
 and coronal MPR reconstruction (B) images showing a diffuse pattern of acute exacerbation.")
 images showing a multifocal pattern of acute exacerbation References: Courtesy of L.Calandriello, Catholic University, Rome.")
 (A) and a pulmonary artery/aorta ratio>1. A minimal pericardial effusion is also seen (arrow in B).")
 at the level of the left atrium revealed a right pneumothorax (asterisk) and pneumomediastinum (yellow arrows). Follow-up chest HRCT scan (B) revealed resolution of the pneumothorax. The patient is candidate to lung transplantation.")
 at baseline and axial HRCT image (B) after two months demonstrates spontaneous left pneumothorax and acute exacerbation. The patient died.")
 and bone window setting (B) show numerous small punctate and branching dense opacities, mainly in fibrotic lung. Oblique MIP projection (C) demonstrates the branching nature of the heterotopic bone formation within areas of fibrosis.")