INTRODUCTION
The term “phakomatoses” (after the word “phakos”,
meaning birthmark) was formulated by the ophthalmologist Van der Hoeve when he described the retinal hamartomas.
Phakomatoses or neurocutaneous syndromes are a heterogeneous group of congenital disorders with variable degree of penetration,
primarily involving structures derived from the embryological neuroectoderm.
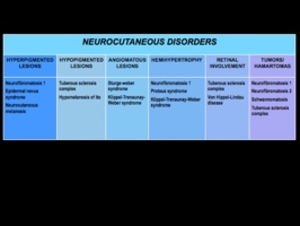
Fig. 1
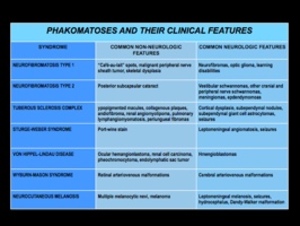
Fig. 2
NEUROFIBROMATOSIS
Neurofibromatosis Type I
Neurofibromatosis type 1 (NF1) also known as Von Recklinghausen’s disease is an autosomal dominant disorder and represents the most common type of
neurocutaneous syndrome.
This disorder is associated with neoplasms of the central and peripheral
nervous systems(optic gliomas,
astrocytomas,
and plexiform and solitary
neurofibromas) and skin (“café-au-lait" spots,
axillary and inguinal freckling).
NF1 is also associated with meningeal and skull dysplasias,
as well as
hamartomas of the iris (Lisch nodules).
Incidence: 1/4000 to 1/25000
Neurofibromas (benign peripheral tumors of Schwann cells and fibroblasts)
are usually assymptomatic,
however may produce compressive radiculopathy
or neuropathy
Other features: hydrocephalus,
scoliosis,
short stature,
epilepsy,
mental
retardation,
pseudoarthrosis of the tibia
MRI: In addition to neoplasms of the central nervous system (astrocytomas,
grades I to IV,
and optic gliomas),
nonneoplastic lesions can occur in the white matter that have high signal on T2-weighted imaging.
They can be solitary or multiple.
Lesions occur in the cerebral and/or cerebellar white matter,
basal ganglia,
and brainstem and can increase in size in the first decade and then usually regress and resolve by the third decade.
Genetics: mutations involving the neurofibromin gene on chromosome 17q11.2,

Fig. 3
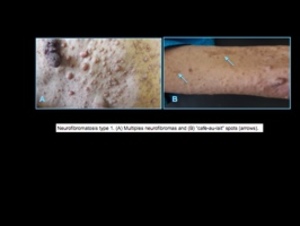
Fig. 4
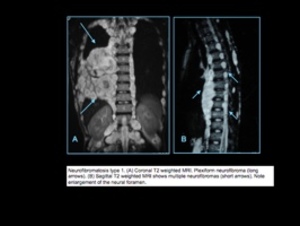
Fig. 5
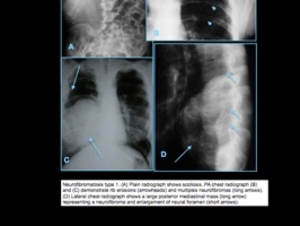
Fig. 6

Fig. 7

Fig. 8
Neurofibromatosis Type II
The incidence of neurofibromatosis type II (NF2) is 1/ 37,000 to 1/50,000
newborns.
NF2 is an autosomal dominant disease involving a gene at chromosome 22q12.
In addition to schwannomas,
patients with NF2 can also have multiple meningiomas and ependymomas.
Age at presentation is 22 to 72 years (mean age = 46 years) with a peak incidence is in the fourth to sixth decades
In NF2,
many patients present in the third decade with bilateral vestibular schwannomas.
Bilateral vestibular schwannomas in 90% of cases which usually results in progressive unilateral deafness
Predisposed to meningiomas,
gliomas,
schwannomas of cranial and spinal nerves
MRI: circumscribed or lobulated lesions with low-intermediate signal on T1-weighted imaging and high signal on T2-weighted imaging and fat-suppressed T2-weighted imaging,
usually showing prominent gadolinium contrast
enhancement that can be heterogeneous in large lesions due to cystic
degeneration and/or hemorrhage.
CT: circumscribed or lobulated lesions,
intermediate attenuation,
with contrast enhancement.
Large lesions can have cystic degeneration and/or hemorrhage.
Other lesions: skin lesions are seldom found,
juvenile posterior subcapsular lenticular opacity
Genetics: NF2 gene on chromose 22q codes for neurofibromin 2,
schwannomin or merlin (part of family of cytoskeletal proteins)
Another syndrome with multiple schwannomas is schwannomatosis (incidence from 1/40,000 to 1/1.7 million) in which patients have multiple schwannomas without involvement of cranial nerve VIII and the peak age of incidence is between 30 and 60 years.
It is related to germline mutation of the SMARCB1 gene (also known as the INI tumor suppressor gene) on chromosome 22.
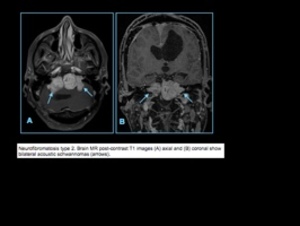
Fig. 9
LEGIUS SYNDROME
Legius syndrome is an autosomal dominant condition characterized by “cafe-au- lait” spots.
It was first described in 2007 and is often mistaken for neurofibromatosis type I (NF-1).
It is caused by mutations in the SPRED1 gene.
It is also known as neurofibromatosis type 1-like syndrome (NFLS).
Other signs and symptoms of Legius syndrome may include an abnormally large head (macrocephaly) and unusual facial characteristics.
Although most people with Legius syndrome have normal intelligence,
some affected individuals have been diagnosed with learning disabilities,
attention deficit disorder ir attention deficit hyperactivity disorder.
Tuberous Sclerosis
Tuberous sclerosis also known as Bourneville’s disease is an autosomal dominant disorder associated with hamartomas in multiple organs.
Extraneural lesions include cutaneous angiofibromas (adenoma sebaceum),
subungual fibromas,
visceral cysts,
renal angiomiolipomas,
intestinal polyps,
cardiac rhabdomyomas,
and pulmonary lymphangioleiomyomatosis.
Cortical hamartomas (tubers),
subcortical glioneuronal hamartomas,
subependymal glial hamartomas (nodules),
and subependymal giant cell
astrocytomas are nonmalignant lesions associated with tuberous sclerosis.
Incidence: 1/29,000
Characteristics: cutaneous lesions,
seizures,
mental retardation
Skin lesions: adenoma sebaceum,
ash leaf-shaped hypopigmented
macules,
shagreen patches (yellowish thickenings in lumbosacral region),
depigmented nevi
Neural lesions: subependymal nodules which may be calcified are characteristic
predisposed to ependynomas and childhood astrocytomas (90% are
subependymal giant cell astrocytomas)
MRI: cortical-subcortical lesions (tubers) usually have high signal on T1-weighted imaging and low signal on T2-weighted imaging in neonates and
infants; changes to low-intermediate signal on T1-weighted imaging and high signal on T2-weighted imaging in older children and adults.
Calcifications occur in 50% in older children,
and gadolinium contrast
enhancement is uncommon.
Subependymal giant cell astrocytomas usually do not have restricted diffusion.
Magnetic resonance spectroscopy: subependymal giant cell astrocytomas can have elevated choline and reduced N-acetylaspartate (NAA).
CT: cortical-subcortical lesions (tubers) have variable attenuation.
Calcifications are seen in 50% of older children
Genetics: caused by mutations of the TSC1 gene on 9q or the TSC2 gene
on 16p code for tuberins which modulate GTPase activity
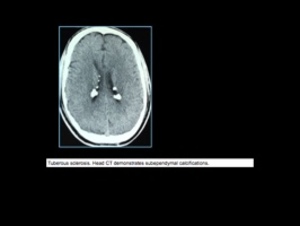
Fig. 10

Fig. 11
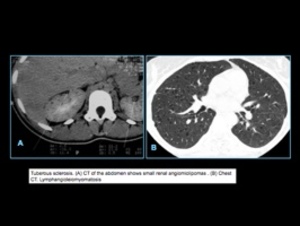
Fig. 12
Von Hippel-Lindau DISEASE
In Von Hippel Lindau (VHL) disease,
multiple hemangioblastomas involving the central nervous system occur,
as well as clear-cell renal carcinoma,
pheochromocytoma,
endolymphatic sac tumor,
neuroendocrine tumor,
adenoma of the pancreas,
and epididymal cystadenoma.
VHL disease occurs in adolescents and young and middle-aged adults.
Incidence: 1/40,000
Characteristics: retinal angiomas and cerebellar hemangioblastomas
These hemangioblastomas may produce erythropoietin and result in
polycythemia.
They account for 1–3% of intracranial neoplasms and
usually occur in middle-aged adults and rarely in children,
except for
patients with VHL disease.
Other lesions: hemangioma of spinal cord,
cysts of kidney,
pancreas,
epididymis,
liver
Genetics: Tumors occur as sporadic mutations of the VHL gene or as an
autosomal dominant germline mutation of the VHL gene on chromosome
3p25–26 resulting in VHL disease.
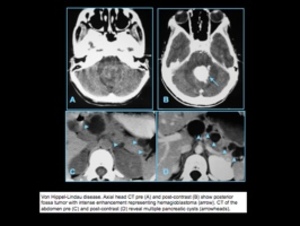
Fig. 13
STURGE-WEBER SYNDROME
Also known as encephalotrigeminal angiomatosis,
Sturge-Weber syndrome is a non-inherited sporadic neurocutaneous syndrome associated with ipsilateral port wine cutaneous lesion,
seizures,
glaucoma,
and hemiparesis.
Results from persistence of primitive leptomeningeal venous drainage
(pial angioma) and developmental lack of normal cortical veins,
producing chronic venous congestion and brain ischemia.
Patients often present with progressive neurologic impairment.
MRI: prominent,
localized,
leptomeningeal contrast enhancement,
usually in parietal and/or occipital regions in children
Unilateral in 80%,
bilateral in 20%
Low signal on T2-weighted imaging is often seen at the involved gyri due to
dystrophic calcifications,
gyral gadolinium contrast enhancement,
localized
atrophic changes in brain adjacent to the pial angioma,
Gyral calcifications > 2 years,
progressive cerebral atrophy in region of pial
angioma,
enlarged ipsilateral choroid plexus
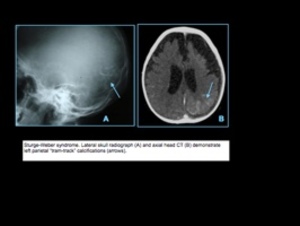
Fig. 14
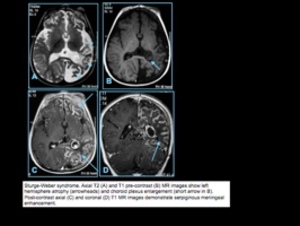
Fig. 15
WYBURN-MASON SYNDROME
Wyburn-Mason syndrome is a neurocutaneous disorder characterised by
arterio-venous malformations (AVMs) in the brain and retina an cutaneous nevi.
Multiple AVMs can be seen in Rendu-Osler-Weber syndrome and Wyburn-Mason syndrome.
NEUROCUTANEOUS MELANOSIS
Neurocutaneous melanosis or melanocytosis is a rare nonfamilial disorder with focal and/or diffuse proliferation of melanocytes in leptomeninges associated with large and/or numerous cutaneous nevi.
This disease presents in infants and young children and rarely occurs in adults.
Cutaneous nevi are typically benign.
Melanocytes in the leptomeninges change to CNS melanoma in 40 to 50% of cases in patients with neurocutaneous melanosis.
Meningeal melanocytoma is a benign rare pigmented tumor consisting of
leptomeningeal melanocytes which typically occur in the posterior cranial fossa or spinal canal in patients with a mean age of 42 years.
MRI: intra-axial lesions usually < 3 cm in brain parenchyma/brainstem (anterior temporal lobes,
cerebellum,
thalami,
inferior frontal lobes) that show
intermediate-slightly high signal on T1-weighted imaging secondary to
increased melanin and decreased signal on T2-weighted imaging,
which present gadolinium contrast enhancement.
Leptomeningeal lesions have irregular margins,
intermediate and/or high signal on T1- weighted imaging in the sulci,
intermediate-slightly high signal on T2-weighted imaging,
high signal on FLAIR,
and leptomeningeal gadolinium contrast enhancement.
CT: may show subtle hyperdensity secondary to increased melanina,
Other features: hydrocephalus,
vermian hypoplasia,
arachnoid cysts,
Dandy-Walker malformation.

Fig. 16
OSLER-WEBER-RENDU
Osler-Weber-Rendu disease is a rare autosomal dominant disorder that affects blood vessels throughout the body (causing vascular dysplasia) and results in a tendency for bleeding.
It is a multiorgan pathology with a prevalence of 1 in 10,000 to 1 in 5000 and characterized by the presence of multiple small telangiectases of the skin,
mucous membranes,
gastrointestinal tract,
and other organs,
with associated recurrent episodes of bleeding from affected sites.
Recurent and severe epistaxis is the most common presentation,
frequently leading to severe anemia that necessitates transfusion.
Gastrointestinal bleeding is also prevalent.
Symptom onset may be delayed until the fourth decade of life (~90% of patients manifest by age 40 years) or later.
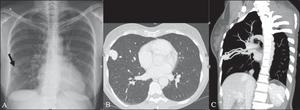
Fig. 17: A: Posteroanterior chest radiography showing nodule with soft-tissue density in the right lung (arrow). B: Axial CT section, with maximum intensity projection (MIP) showing nodule with soft-tissue attenuation in the middle lobe of the right lung. C: Oblique MPR reconstruction demonstrating the vascular nature of the previously described nodule, with at least two pulmonary arterial branches supplying a malformative nidus and main pulmonary vein drainage (pulmonary arteriovenous malformation.
References: Agnollitto P, Barreto A, Barbieri R et al. (2013) Rendu-Osler-Weber syndrome: what radiologists should know. Literature review and three cases report. Radiologia Brasileira vol.46 no.3
















