ECR 2016 / C-0508
Spinal involvement in mucopolysaccharidoses: what radiologists need to know
This poster is published under an open license. Please read the disclaimer for further details.
Congress:
ECR 2016
Poster Number:
C-0508
Type:
Educational Exhibit
Keywords:
Connective tissue disorders, Congenital, Imaging sequences, MR, Digital radiography, CT, Musculoskeletal spine, Musculoskeletal system, Musculoskeletal bone
Authors:
A. Semprini1, A. Leone2, L. Tonetti1, V. Zecchi2, M. Marino1, C. Colosimo2; 1Roma/IT, 2Rome/IT
DOI:
10.1594/ecr2016/C-0508
 Spinal Involvement in mucopolysaccharidoses: a review. Childs Nerv Syst 31:203-212.")
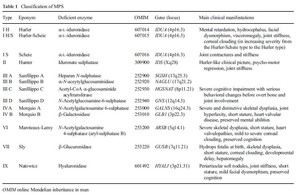
Table 1:
Classification of MPS.