Smoking related lung diseases are the respiratory manifestations of diseases that are related to smoking.
Smoking affects the lungs in numerous ways,
and can be classified under the following headings:
Smoking related interstitial lung diseases (SR-ILD), including entities secondary to smoking-related lung inflammation such as:
- Respiratory Bronchiolitis (RB)
- Respiratory Bronchiolitis-Interstitial Lung Disease (RB-ILD)
- Desquamative Interstitial Pneumonia (DIP)
- Pulmonary Langerhans Cell Histiocytosis (PLCH)
- Acute eosinophilic pneumonia
as well as chronic fibrosing lung diseases strongly associated with cigarette smoke including
- Idiopathic pulmonary fibrosis (IPF)
- Combined pulmonary fibrosis and emphysema (CPFE)
Respiratory bronchiolitis (RB) is an inflammatory process of small airways.
It refers to a histological finding that can be often seen in heavy smokers.
Clinical presentation:
It consists of mild chronic inflammation and accumulation of pigmented macrophages within respiratory bronchioles and related alveoli.
It is usually asymptomatic and of little clinical significance.
It is closely related to respiratory bronchiolitis interstitial lung disease (RB-ILD) which represents a more advanced form of the same condition and is also seen almost exclusively amongst smokers.
HRCT Features:
Usually respiratory bronchiolitis has no imaging findings,
although occasionally minor patchy ground glass opacities and ill-defined centrilobular nodules may be seen,
which tend to be more pronounced in the upper zones.
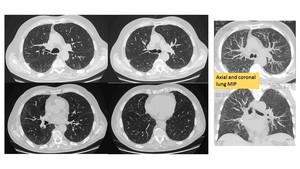
Fig. 1: Mild shortness of breath over many months in a heavy smoker. No fever and no sputum. High-resolution CT image obtained through the upper lungs showed bilateral centrilobular ill defined micro-nodules. This was in keeping with a diagnosis of respiratory bronchiolitis.
Respiratory bronchiolitis interstitial lung disease (RB-ILD) is a smoking related interstitial lung disease closely related to respiratory bronchiolitis,
but demonstrating more severe histological,
imaging and clinical findings.
RBILD is characterized by the presence of pigmented macrophages and mild interstitial inflammatory changes centring on respiratory bronchioles and neighbouring alveoli.
Clinical presentation:
RB-ILD is typically associated with heavy smoking (usually of 30 pack-years or more) and is often seen in young middle-aged patients (30-40 years of age).
There is a slight male predominance.
Presentation tends to be with progressive shortness of breath and chronic cough.
Inspiratory crackles are heard in up to half of patients.
Clubbing is rare.
Patients with RBILD have a generally good prognosis,
particularly with smoking cessation.
HRCT features:
- Ground glass opacities: may have a slight upper zone predilection,
- Poorly defined centrilobular nodules,
- If advanced,
fibrosis may be evident typically affecting the subpleural regions and more so in the lower lung zones
- Patchy areas of hypoattenuation (~40%) with a lower lung predominance,
- Other changes related to smoking:
- bronchial wall thickening: can be central +/- peripheral
- centrilobular emphysema.

Fig. 2: 65 year-old-male, a lifelong heavy smoker. Axial HRCT images, MIP and MINIP reformarts showed multiple small and ill-defined nodules and patchy ground-glass opacities in both lungs. Bronchial washings returned a dark stained bronchial lavage, with heavily pigmented macrophages. Diagnosis: RB-ILD.

Fig. 3: HRCT of a 51-yr-old male, strong smoker, with respiratory bronchiolitis-associated interstitial lung disease showing patchy areas of ground-glass opacifications with bronchiolocentrci distribution, accentuated in the upper part of the lung.
Desquamative interstitial pneumonia (DIP) is an interstitial pneumonia closely related to,
and thought to represent the end stage of respiratory bronchiolitis interstitial lung disease (RB-ILD).
Desquamative interstitial pneumonia is an uncommon disorder characterised by macrophage accumulation within the alveoli.
Clinical presentation:
DIP occurs most commonly in patients between 30 and 50 years of age,
approximately 90% of which is current cigarette smokers.
It is more common in men than in women.
It may manifest with dyspnoea or cough.
The prognosis is good,
improving with smoking cessation and corticosteroids therapy.
In some rare cases the progression of disease may lead to fibrosis.
HRCT Features:
DIP is usually characterised by diffuse ground-glass opacities,
which correlate histologically with the spatially homogeneous intra-alveolar accumulation of macrophages and thickening of alveolar septa:
- Bilateral and symmetric (86%)
- Basal and peripheral (60%)
- Patchy (20%)
- Diffuse (20%)
Other frequent CT findings include spatially limited,
irregular linear opacities and small cystic spaces,
which are indicative of fibrotic change (50% of patients).
Other changes related to background smoking-related lung disease are often seen: bronchial wall thickening and centrilobular emphysema.
Despite differences in the CT appearance of RB-ILD and DIP,
imaging findings may overlap and may be indistinguishable from each other.
To improve diagnostic accuracy,
lung biopsy is required in all cases of suspected RB-ILD or DIP.
Honeycombing may be seen in less than one-third of cases,
and if present tends to be usually peripheral and limited in extent.

Fig. 4: DIP in a 55-year-old man with a 30 pack-year history of smoking who presented with a cough, gradually increasing shortness of breath, and mild restriction at pulmonary function testing, with diffusing capacity of the lung for carbon monoxide (Dlco) 50% of the predicted.
High-resolution CT image showed bilateral diffuse ground-glass opacity, peripheral reticulation, and small cysts (the yellow arrows).

Fig. 5: 45-year-old patient affected by DIP. HRCT showed signs of progression of disease to fibrosis, which is an infrequent finding.
High-resolution CT image obtained through the upper lungs showed paraseptal and centrilobular emphysema. Coronal reformatted image showed the basilar predominant distribution of the ground-glass opacity, the traction bronchiectasis, and the apical emphysema.

Fig. 6: DIP in a 50-year-old woman with a 25 pack-year history of smoking. High-resolution CT image obtained through the right lower lung at initial diagnosis showed diffuse ground-glass opacity.
High-resolution CT image obtained through the right lower lung 2 years later showed near-complete resolution of the ground-glass opacity.
Pulmonary Langerhans cell histiocytosis (PLCH) can be seen as part of widespread involvement in patients with disseminated Langerhans cell histiocytosis (in which Langerhans cell accumulations involve one or more body systems,
including bone,
lung,
pituitary gland,
mucous membranes,
skin,
lymph nodes,
and liver),
or more frequently as a distinct entity in young adult smokers.
Clinical presentation:
PLCH is diagnosed predominantly in younger adults,
during the third or fourth decade of life.
PLCH has no sex-based predominance,
the most striking epidemiological characteristic of adult PLCH is that 90–100% of patients are smokers,
often smoking > 20 cigarettes per day.
No other epidemiological factor associated with PLCH has been identified.
The main respiratory symptoms are dry cough and dyspnoea on exertion,
which can be associated with nonspecific constitutional manifestations such as asthenia,
fever,
night sweats and weight loss.
Bone lesions (< 20% of patients),
diabetes insipidus with polyuria and polydipsia and skin lesions are the most common extrapulmonary manifestations.
Complete remission of PLCH may occur on smoking cessation.
Spontaneous pneumothorax (responsible for chest pain) leads to the diagnosis in ∼10–20% of cases.
Pneumothorax requires drainage.
In a minority of patients (~20%) and more frequently in those who continue to smoke,
the disease is progressive with deterioration in respiratory function and eventual end-stage pulmonary fibrosis and the only therapeutic option is represented by the lung transplantation.
HRCT Features:
High-resolution CT is sensitive and specific for the diagnosis of PLCH.
The HRCT appearances of PLCH vary according to the chronicity of the disease.
In the early stages,
nodules (which correspond with Langerhans cell granulomas) are the predominant features,
while cysts tend to develop later.
It has been shown that cysts in PLCH probably arise because of focal dilatation of bronchi caused by destruction of small airway bronchial walls due to Langerhans cell lesions.
Cysts can be identified in only 1-15% of cases ,
and range from 1-3 cm in diameter.
Distribution is the key in differentiating PLCH from other cystic lung diseases with predilection for the mid and upper zones and regional sparing of the costophrenic recesses,
anterior right middle lobe and lingula left upper lobe.
However,
the combination of nodules,
cavitating nodules and cysts in a smoker should allow a confident and accurate diagnosis to be made on CT alone.
There is a preservation of lung volumes or even hyperinflation.
Reduced lung volumes are uncommon and only seen in end-stage fibrotic cases.
- Nodules
- more pronounced early in the disease
- may range in number from a few to innumerable
- 1-10 mm in diameter (typically 1-5 mm)
- centrilobular distribution may also be peribronchial or peribronchiolar
- usually have irregular margins
- may be cavitatary nodules with thick walls,
later becoming cysts
- surrounding lung parenchyma appears normal
- Cysts
- more pronounced later in the disease
- usually less than 10 mm in diameter
- may measure up to 2-3 centimetres in size
- the extreme bases may be preserved
- usually thin-walled,
but on occasion may be up to a few millimetres thick
- confluence of 2 or more cysts results in bizarre shapes
- bilobed
- cloverleaf
- branching
- internal septations
Other common findings include:
- Ground-glass and/or reticular opacities
- Mosaic attenuation
- Septal line thickening
- Emphysema
In late disease,
other findings include:
- Cyst coalescion
- Fibrosis
- Honeycombing
The appearance of new nodules later in the disease (when cystic change is established) indicates disease progression but is a rare finding.
Complications:
- Cyst rupture
- Spontaneous pneumothorax: may be the first presentation
- Pneumomediastinum
- Interstitial fibrosis
- Pulmonary arterial hypertension and cor pulmonale
- End-stage pulmonary fibrosis and respiratory failure

Fig. 7: PLCH in a 30-year-old man with a 40 pack-year history of smoking who presented with a cough and dyspnea, a Dlco 45% of the predicted, and restrictive Pulmonary Functional Tests (PFT) results. High-resolution computed tomography scans of upper and middle lung regions showed multiple thin-walled cysts, some with irregular shapes (arrows), in both lungs. Multiple nodules (short arrows), some of which showed cavitation, were also present. MINIP reconstruction on coronal plane demonstrated upper lobar predominance.

Fig. 8: HRCT of a 27 y-old smoker with advanced PLCH. Multiple cysts were present of varying sizes and shape. Note is again made of upper lobe predominance. The patient had osteolityc lesion of the skull (red arrow) and skin lesions (granulomas).

Fig. 9: HRCT scan of 31 year-old male with PLCH, complicated by extensive pneumothorax.
Acute eosinophilic pneumonia (AEP) is a rare syndrome characterized by abrupt onset of fever,
dyspnea and cough along with a characteristic radiographic pattern which includes diffuse bilateral pulmonary infiltrates.
AEP is a serious cause of acute hypoxemic respiratory failure in young adults and is potentially life threatening.
The etiology of AEP is not well understood,
however several studies have reported smoking as a trigger for AEP.
Peripheral blood eosinophil counts are usually normal,
although they can become elevated during the subsequent clinical course.
A very high eosinophil count in bronchoalveolar lavage is characteristic of the condition.
Clinical presentation:
- Youg adults
- Smokers
- Fever
- Dyspnea
- Cough
- Eosinophilia of more than 25% in BAL cytology
HRCT Features:
- Bilateral ground-glass areas: common
- Interlobular septal thickening: common
- Pleural effusions: can be present in ~80% (range 60-100%) of cases
- Thickening of bronchovascular bundles: present in around two-thirds of cases
- Air-space consolidation: present in around half of cases
- Ill-defined centrilobular nodules: present in around one-third of cases
- Distribution: random

Fig. 10: 35 year-old female, with a long history of smoking. Cough, weight loss, and low grade fever. X-rays showed bilateral air space opacities that are scattered throughout both lungs.
HRCT axial and coronal images showed bilateral areas of ground glass opacities and consolidations at the subpleural and peribronchial regions, more at the upper and middle zones.
Bronchoalveolar lavage showed significant eosinophilia.
Follow-up x-ray after 10 days of systemic low dose corticosteroids showed interval resolution of the previously reported air space opacities in both lungs.
Idiopathic pulmonary fibrosis (IPF) is defined as a “specific form of chronic,
progressive,
fibrosing interstitial pneumonia of unknown cause,
occurring in adult lungs and associated with the histopathological and/or radiological pattern of Usual Interstitial Pneumonia (UIP)”.
A relationship between cigarette smoking and IPF is recognized.
IPF disease progression is variable,
irreversible,
and inevitably fatal.
Lung transplantation is the olny therapeutic option,
but it is available for a small number of patients with IPF
Clinical Presentation:
- Male> female
- 65 years old
- Smoker or former smoker
- Dry cough
- Dyspnea at exertion
- Basal inspiratory crackles on auscultation
- 3-5 years survival after diagnosis
- Lung failure
Comorbidities may include
- Pulmonary hypertension
- Gastro-oesophageal reflux
- Emphysema
The clinical course is that of gradual deterioration and the condition carries a rather poor prognosis with median survival ranging from 2.5 to 3.5 years from the time of diagnosis.
Some reports have suggested a slowing of progression with treatment by pirfenidone or nintedanib.
Diagnostic criteria:
Multidisciplinary approach in tertiary setting is strongly advised.
Contributions from pulmonists,
chest radiologists,
and chest pathologist are crucial reaching the correct diagnosis of IPF.
ATS/ERS (American Thoracic Society and European Respiratory Society) has developed major and minor criteria for the diagnosis of IPF in the absence of a surgical lung biopsy:
Major criteria
- Exclusion of other known causes of interstitial lung disease (toxic effects of certain drugs,
environmental exposures,
connective tissue diseases)
- Abnormal results of pulmonary function studies,
including evidence of restriction and impaired gas exchange
- Definite UIP pattern on HRCT chest
- Transbronchial lung biopsy or bronchoalveolar lavage shows no features to support an alternative diagnosis
Minor criteria
- Age >50 years
- Insidious onset of otherwise unexplained dyspnea on exertion
- Duration of illness being over 3 months
- Bibasilar inspiratory crackles (dry or “Velcro” type)
HRCT Features:
High-resolution CT has been shown to be a highly accurate tool for diagnosis of UIP,
with a positive predictive value of 95%–100%
The need of surgical biopsy is reduced after HRCT (40%).
UIP-pattern of fibrosis is is characterized by:
- Irregular reticular opacities,
with intralobular distribution
- No relevant ground-glass
- Honeycombing +/- “traction” bronchiectases with non segmental distribution and volume loss is the strongest CT indicator of UIP
- Upper lobe involvement is characteristic but less severe than that in the lower lobes
- Distribution is bilateral,
basal and peripheral,
but sometimes patchy or asymmetrical (30%)
In a subgroup of patients the imaging finding of UIP overlap with NSIP and biopsy may be necessary to obtain the correct diagnosis.
Complications:
PNEUMOMEDIASTINUM: 5%
PNX: 6%
Lung Cancer: 10% of the pts with UIP develop lung cancer (mainly squamos cell type); the likelihood to develop lung cancer in pts affected by IPF increases with the age and the duration of the follow-up.

Fig. 11: Advanced IPF in a 69-year-old former smoker with severe hypoxemia at rest.
Irregular reticular opacities, with intralobular distribution.
No relevant ground-glass.
Honeycombing +/- “traction” bronchiectases with non segmental distribution and volume loss was the strongest CT indicator of UIP.
Upper lobe involvement was characteristic but less severe than that in the lower lobes.
Propeller-blade distribution from the base to the apex.

Fig. 12: HRCT shows all the features of UIP pattern in a 70 year-male ex smoker, affected by IPF: the typical distribution of honeycombing (bilateral, postero-basal and peripheral), the diffuse reticular opacities, the “traction” bronchiectasis.
Note the prone and supine acquisition, and the multiplanar reformats images (MPR).

Fig. 13: Traction bronchiectasis can mimick honeycombing; but the presence of bronchiectasis can be highlighted by the MINIP reconstruction intensity projection).Volume rendering reformats have an iconoclastic value.
Combined pulmonary fibrosis and emphysema (CPFE) are a possible new addition to a growing list of smoking-related lung disease characterized by the coexistence of emphysema and pulmonary fibrosis in the same patient.
Clinical Presentation:
- Tobacco smoking
- Severe dyspnoea
- Hypoxaemia on exercise
- Normal spirometry and lung volumes in the setting of severely impaired gas exchange
- This syndrome frequently is complicated by pulmonary hypertension,
acute lung injury,
and lung cancer
- Patients with CPFE syndrome have worse survival and higher mortality rate than patients with pulmonary fibrosis alone
HRCT Features:
- Centrilobular and/or paraseptal emphysema: often upper zone predominant
- Pulmonary fibrosis of the lower lobes: can be of UIP or NSIP pattern

Fig. 14: CPFE in a 65-year-old male with an 80 pack-year history of smoking, presenting with cough, dyspnea, resting hypoxemia, and clubbing; a Dlco 35% of the predicted; and a family history of IPF (a brother).
HRCT showed bilateral upper lobe paraseptal emphysema with bullae.
High-resolution computed tomogram of the same patient showed lower lobe subpleural honeycombing, fibrosis, and traction bronchiectasis.




 50% of the predicted.
High-resolution CT image showed bilateral diffuse ground-glass opacity, peripheral reticulation, and small cysts (the yellow arrows).")


 results. High-resolution computed tomography scans of upper and middle lung regions showed multiple thin-walled cysts, some with irregular shapes (arrows), in both lungs. Multiple nodules (short arrows), some of which showed cavitation, were also present. MINIP reconstruction on coronal plane demonstrated upper lobar predominance.")
 and skin lesions (granulomas).")



, the diffuse reticular opacities, the “traction” bronchiectasis.
Note the prone and supine acquisition, and the multiplanar reformats images (MPR).")
.Volume rendering reformats have an iconoclastic value.")
.
HRCT showed bilateral upper lobe paraseptal emphysema with bullae.
High-resolution computed tomogram of the same patient showed lower lobe subpleural honeycombing, fibrosis, and traction bronchiectasis.")
