Biliary diseases in pediatric patients have been summarized as proposed in Table 2
Congenital biliary diseases include biliary atresia,
cystic biliary lesions,
agenesia/malformation of gallbladder.
Acquired biliary diseases include lithiasis,
infections and Primary Sclerosing Cholangitis.
Biliary Atresia
Biliary atresia is the most common cause of persisting neonatal jaundice which affects 1 person in every 8,000,
occurring,
in particular,
in Asian populations.
The disease – in its typical evolution – is characterized by cholestatis and development of cirrhosis and hepatic fibrosis as a result of the progressive fibrosis of the biliary tree [2].
Asplenia or polysplenia,
cardiac abnormalities,
situs inversus,
symmetric bilobate liver and absent or interrupted inferior vena cava may be often associated with biliary atresia.
Among cholestatic symptoms,
biliary atresia may develop jaundice but also acholic stools,
coloured urine,
itching (after the first month) and hepatomegaly.
A clear clinical finding is represented by the increase of conjugate bilirubin [3].
Three kinds of anatomic variants have been proposed for biliary atresia – according to the Ohi classification system,
which has been adopted by the Japanese Biliary Atresia Registry.
The type I can be corrected by means of a surgical intervention.
This variant is known as atresia of the CBD – with no involvement of right,
left and common hepatic ducts.
Type II identifies the atresia of the hepatic duct,
whereas type III refers to the extra-hepatic and intra-hepatic bile ducts of the porta hepatis [2].
Moreover,
biliary atresia can be further classified taking into consideration the level of atresia and the porta hepatis ducts morphology showing,
for instance,
bile lake or dilatation.
The diagnosis of biliary atresia can be provided by liver biopsy; however,
intra-operative cholangiography is considered the gold standard in confirming the disease [2].
Surgical intervention involves the Kasai procedure,
which is generally performed when the newborn is 2 months old.
The intervention is crucial since life expectancy amounts to just 18 months when surgical repair is not performed.
The Kasai procedure can be decisive with patients affected by the type I,
or it can be preparatory for the liver transplant.
The intervention inolves the excision of the atretic porta hepatis bile ducts.
A Roux-en-Y hepaticojejunostomy follows.
After three months from surgical repair,
the total bilirubin concentration must be under 2.0 mg/dL,
establishing a proper biliary drainage and cancelling the need for a liver transplant.
Furthermore,
a successful Kasai procedure is pivotal in preventing the patient suffering from chronic cholestasis Table 3 [4].
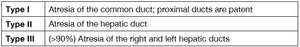
Table 3: Table 3. Kasai Classification.
References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital
Figures Fig. 2 ,
Fig. 3 ,
Fig. 4 and Fig. 5 describe the different types of Kasai Classification.
Ultrasound is the first technique used for these babies with symptoms of cholestasis,
namely to make a differential diagnosis from choledochal cysts.
US findings in biliary atresia could be a normal echo texture of the liver or – in case of advanced disease – the presence of cirrhosis or portal venous hypertension.
Typically US imaging finding Fig. 6 depicted in biliary atresia is the "triangulard cord sign",
that is a hyperechoic area of fibrous tissue localized in the porta hepatis,
with a triangular shape [5].
It has been demonstrated that a ‘cord’ measuring more than 4 mm in thickness has about 80% of sensitivity and 98% of specificity for the diagnosis of biliary atresia.
Also the ‘gallbladder ghost triad’ is an important clue for the diagnosis,
including an atretic gallbladder (less than 1.9 cm in length),
with a lobular contour and a thin mucosa.
The gallbladder may also be not recognizable,
absent or completely normal [5,
6].
US Color-Doppler (Figure 6) is useful in the diagnosis; a larger right hepatic artery is frequently encountered in pediatric patients together with hepatitis and biliary atresia.
More in detail,
a value of 1.5 mm for the diameter of the hepatic artery – and a value of 0.45 for hepatic artery/portal vein ratio are also depicted in biliary atresia.
Moreover,
the Color-Doppler could help radiologists to avoid a common false-negative: the hepatic artery may be confused with a normal common bile duct [7].
MRCP is a second step in the diagnosis of biliary atresia,
with the presence of signs very similar to those resulting from the US procedure.
The equivalent of the ‘triangular cord sign’,
seen with US,
is a fibrous area,
with the same shape,
hypointense on T1 and hyperintense on T2,
localized in the porta hepatis.
The absence of the extra hepatic bile ducts and a small or absent gallbladder are also confirmed with MRCP [8] ( Fig. 7 ,
and Fig. 8 ).
Congenital cystic lesions of the biliary tree
Radiologists should provide a differential diagnosis between biliary atresia and congenital cystic lesions of the biliary tree; as previously described,
biliary atresia requires prompt surgical intervention in order to avoid the risk of biliary cirrhosis.
Congenital cystic lesions include malformation of biliary system – which may involve intra- or extra-hepatic ducts.
These abnormalities can be surgically operated on in older patients as well,
according to the symptoms.
Choledochal cysts is a rare pathology generally occurring in 1/100,000 people in Western countries; a peak of incidence has been reported in Asian populations: its occurrence rises to 1/13,000 in Japan.
The disease is more commonly found among women.
[2].
The etiopathogenesis is multifactorial since it can have a congenital feature and an acquisitive part.
The latter depends on the abnormal pancreaticobiliary junction (APBJ) and consists of the early convergence of the common bile duct (CBD) with the main pancreatic duct (Wirsung duct).
The early convergence can lead to the exposition of the biliary epithelium to pancreatic enzymes.
Therefore,
when APBJ occurs there is a “long common channel” that is the union of the main pancreatic duct to the CBD with more than 15 mm from the ampulla of Vater.
Namely,
the APBJ can also exist without the choledocal cysts but in more than 96% of cases the choledocal cysts is associated with APBJ [9].
Table 4 [2],[10].
The most common symptoms include abdominal pain,
jaundice,
and – in some cases – a palpable mass in right upper quadrant.
Other possible manifestations are cholangitis,
signs of portal hypertension,
liver dysfunctions and pancreatitis.
The type I is the most frequent,
immediately followed by type IV; in both cases,
surgical excision is highly recommended [11].
US visualization of choledochal cyst is based on a cystic lesion,
localized in the right upper quadrant,
with a connection to the biliary tree.
Biliary cyst should be distinguished from gallbladder Fig. 9 .
MRCP is performed in order to classify the cystic biliary disease according to Todani classification; on MRCP sequences,
it is possible to obtain measurements of cystic lesions and their extension [2] ( Fig. 10 - Fig. 11 - Fig. 12 ).
Caroli disease
Caroli disease is a rare autosomal recessive disease,
characterized by segmental non-obstructive saccular or fusiform dilatation of intrahepatic bile ducts – which may reach up to 5 (cm).
These dilated ducts may contain calculi or sludge.
The pathology can affect just one or both lobes but,
in most cases,
it affects the left one.
Some complications are cholangitis,
cholangiocarcinoma (7%) and cirrhosis; whenever the disease is associated with congenital hepatic fibrosis or with renal disease,
it is called Caroli syndrome [2],
[12].
Main symptoms of Caroli disease include fever,
transient jaundice and abdominal pain.
The therapy consists either of hepatic resection when the disease is localized or,
otherwise,
liver transplant [2].
US findings connected with Caroli disease are some cystic lesion of the intrahepatic bile ducts,
with in their lumen echogenic septas.
These saccular increased ducts surround the small portal vein branches,
creating the characteristic ‘central dot sign’ – which may be seen on enhanced-CT and MR acquisitions.
Sometimes the exact connection between these cysts and the biliary tree could be difficult to identify with the use of US and CT only,
confusing Caroli disease with polycystic liver disease.
MRCP is helpful to distinguish them better [13].
Differential diagnosis include polycystic liver disease,
which is characterized by multiple cystic lesions in the hepatic parenchyma.
More in detail,
autosomal dominant polycystic liver disease may be encountered in isolation or associated with autosomal dominant polycystic kidney disease [12].
Gallbladder Agenesis
The insurgence of gallbladder agenesis is rare,
occurring in 10-65 individuals per 100,000; it is more commonly found in women,
especially aged between 20 and 30 years-old.
The etiology is still unknown but it is generally caused by the lack of cystic bud or absence of vacuolation.
In some cases,
it is possible to think of a genetic component since there are some possible associated diseases – such as abnormalities of the genitourinary tract,
cardiovascular malformations and gastrointestinal anomalies,
polysplenia and asplenia.
Nonetheless,
in 70% of cases,
gallbladder agenesis is an isolated anomaly.
Patients are divided into three groups.
The first group is made up of individuals who do not show any symptoms,
the anomaly is randomly found after the patient’s death thanks to autopsy,
in the second group patients suffer from biliary colic and/or jaundice or dyspepsia.
Concerning this group,
even if there is no gallbladder the patients have symptoms of a biliary colic caused either by a dysfunction of the Oddi sphincter or by calculi in the common bile duct.
The third group consists of children showing also other associated severe fetal anomalies.
US findings in gallbladder agenesis are: nonvisualization of the gallbladder and no dilatation of the intrahepatic biliary ducts Fig. 13 . In some cases the gallbladder could be described as a contracted/ fibrotic organ.
Sometimes – when gallbladder is not found – an intraoperative ultrasound exam could be useful to demonstrate an ectopic location.
If the gallbladder is not visualized on US or CT scan,
MRCP is used,
usually visualizing a little cystic lesion – measuring about 4 mm – nearby the hepatic common duct.
MRCP could also visualize an ectopic gallbladder or other anomalies associated with the biliary tract.
The therapy is usually traditional,
consisting in the dosing of smooth muscle relaxants [14],[15].
Cholelithiasis and choledocholithiasis
Gallstones may be present in patients of any age: in fact,
they may be depicted in prenatal sonography [16].
In these cases,
the etiopathogenesis might be congenital even if,
nowadays,
gallstones are mainly caused by pediatric obesity; in addition,
gallstones may be caused by supersaturation of bile due to excessive cholesterol levels.
Another kind of calculus,
called pigmented stone,
is associated with hemolytic diseases,
such as sickle cell disease,
thalassemia maior,
spherocytosis and hemolytic anemia.
In this case the hemoglobin,
deriving from hemolysis,
degrades itself resulting in an increase of the bilirubin.
The conjugation of bilirubin and calcium causes the formation of pigmented stones.
Other predisposing factors to calculi formation are intestinal malabsorption and total parenteral nutrition [2].
Among the most common complications,
it is possible to observe acute calculus cholecystitis:,
which is caused by the cystic duct obstruction and gallbladder inflammation.
US findings in cholecystitis are wall thickening (>4/5 mm) Fig. 14 and hyperemia of the gallbladder wall with some fluid in the pericholecystic area.
A second complication is represented by the choledocholithiasis.
It is very difficult to identify the calculus due to the artifact of bowel meteorism.
The acute acalculous cholecystitis (AAC) is another common complication observed: it consists of a typical gallbladder inflammation,
with all the classic US findings without calculi.
In 75% of cases the cholelythiasis is asymptomatic – in both kids and adults.
Nonetheless,
it can rarely present these symptoms: right upper quadrant abdominal pain,
nausea,
vomiting and,
occasionally,
fever.
US is the first examination where there is clinical suspicious of gallstones,
with sensitivity and specificity greater than 95%.
US finding is the hyperechoic appearance of the calculus associated with acoustic shadow.
Fig. 15 .
In cases where there is a large stone or gallbladder contraction,
it is possible to have the “wall echo shadow complex”,
created by two parallel arcs,
which are represented by the gallbladder wall and the stone,
separated by a hypoechoic line of bile.
A semiotic sign is the Murphy sign,
with pain in the right upper quadrant during the ultrasound exploration.
Occasionally it is possible to identify calcified stones on a simple x-ray examination.
MRCP has a sensitivity of 97% in the direct visualization of ductal stones,
typically depicted on MR images as hypointense filling defect of biliary ducts Fig. 16- Fig. 17 .
Symptomatic children need to be surgically treated with a laparoscopic or open cholecystectomy; asymptomatic children suffering from sickle cell disease should be operated on in any case.
On the other hand,
a fetus with asymptomatic gallbladder cholelithiasis needs just sonographic follow-up whenever there are no other biliary tract diseases or predisposing gallstone factors [2],[17].
Primary Sclerosing Cholangitis
Primary Sclerosing Cholangitis is a chronic progressive inflammatory disease of the bile ducts that leads to obliteration of the bile ducts,
and fibrosis of the liver with a possible evolution to cholestasis and biliary cirrhosis.
The etiology is still unknown: it has been described an association with ulcerative colitis (70%). Primary Sclerosing Cholangitis is a rare pathology generally occurring in 2/10,000 children.
It could be an asymptomatic deasease but usually it presents itself wit pruritus,
jaundice,
increasing fatigue and abdominal pain.
Cholangiocarcinoma is an extremely rare evolution of Primary Sclerosing Cholangitis in children,
but still a possible one.
MRCP is the first-line imaging test for the diagnosis with findings as multifocal,
intrahepatic bile duct strictures alternating with dilated ones,
giving a beaded appearance.
Periductal enhancement can be seen after intravenous contrast,
indicating an active inflammation.
The only curative treatment is liver transplantation. [2],[18].

 - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital")
. On figures B, white arrow show a small hypoplasic/atretic gallbladder (punctiform hyperintensity on SSFP image). On figure C, intrapancreatic biliary tract is not demonstrable: type 3 of biliary atresia was radiologically suspected. References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital")
. References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital")
. References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital")
; disease was also complicated by a small calculus in the distal portion of choleducus (not visible on images provided).
References: Courtesy of Dr. Michele Figuera – Radiodiagnostic Department, University Hospital "Policlinico-Vittorio Emanuele"")
 - Radiology I Unit, University Hospital")
. References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital")
 - Radiology I Unit, University Hospital")
 demonstrate a small hypointense area centered in the main biliary duct; it corresponds to a small stone, which occurred in a small baby (4 months old). A percutaneous cholecystostomy was first performed; in a second-time (figure C), a percutaneous approach using a microcatheter was applied, removing the small biliary stone. References: Department of Medical-Surgical Sciences and Advanced Technologies (University of Catania) - Radiology I Unit, University Hospital "Policlinico-Vittorio Emanuele"")