The diagnosis of ALGS is established in a proband who meets clinical diagnostic criteria and/or has a heterozygous pathogenic variant in JAG1 or NOTCH2 identified by molecular genetic testing.
The syndrome is due to mutation in the JAG1 gene (20p12) which encodes a ligand of the Nocth signaling pathway. Type 2 is due to mutations in the NOTCH2 gene (1p12).
Transmission is autosomal dominant, but reduced penetrance (up to 50% of cases) and somatic mosaicism (~ 8%) are frequent. The diagnosis is based on the clinical features and the outcome of the liver biopsy which highlights cholestasis and paucity of the intrahepatic bile ducts.
Prominent forehead and pointed chin (giving the face a triangular appearance), deep-set eyes, straight nose are typical features of the disease.
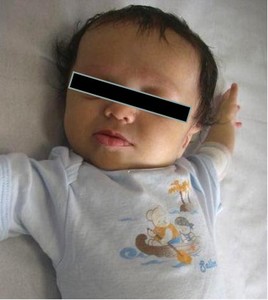
Fig. 1: Typical facial features of Alagille syndrome. Broad forehead, deep-set eyes and pointed.
The severity of liver disease ranges from asymptomatic elevations of liver enzymes to jaundice, chronic cholestasis, and end-stage liver disease. Jaundice and conjugated hyperbilirubinemia may be present in the neonatal period. Increased serum concentrations of bile acids, alkaline phosphatase, gamma-glutamyl transpeptidase, triglycerides, colesterolemia, aminotransferases, coagulopathy are also observed. Impaired bile salt secretion can lead to fat-soluble vitamin deficiencies and malnutrition. Cholestasis manifests as pruritus, increased serum concentration of bile acids, growth failure, and xanthomas.
Screening for ofhthalmic, skeletal, vascular and endocrine (thyroid) abnormalities shoud be performed.
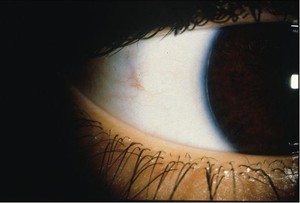
Fig. 2: Posterior embryotoxon involves a thickened and centrally displaced anterior border ring of Schwalbe. The Schwalbe ring represents the junction of the trabecular meshwork with the termination of Descemet’s membrane, and it is visible in 8%–30% of normal eyes as an irregular, opaque ridge 0.5–2.0 mm central to the limbus. The term posterior embryotoxon is used when the Schwalbe ring is visible by external examination. Posterior embryotoxon is usually inherited as a dominant trait. The eye is usually normal but can manifest a number of other anterior segment anomalies that are part of ocular or systemic syndromes,
such as Alagille syndrome (arteriohepatic dysplasia), X-linked ichthyosis, and familial aniridia.
The prognosis is usually good, but complications such as cirrhosis, hemorrhage of varicose veins, refractory ascites and spontaneous bacterial peritonitis may occur. The disease usually stabilizes between 4 and 10 years of age.In the presence of liver failure and heart injury, the risk of death increases, and liver transplant is required.


, X-linked ichthyosis, and familial aniridia.")