BACKGROUND
The pulmonary-renal syndrome is defined by the combination of diffuse alveolar haemorrhage and rapid progressive glomerulonephritis (RPGN) [1,2,11].
Diffuse alveolar haemorrhage comprises the triad of hemoptysis,
diffuse alveolar infiltrates and low hematocrit.
However,
clinical presentation can be variable (slight cough,
progressive dyspnea,
manifest hemoptysis).
Symptoms need not present simultaneously.
Rapid-progressive glomerulonephritis manifests in quick loss of renal function (within a few days/weeks) and the presence of a nephritic sediment with deformed erythrocytes of a glomerular origin and possibly red cell casts [11].
Due to the heterogeneous pathogenesis of small vessels vasculitis,
the entities encompassed in the term "pulmonary-renal syndrome" can be classified based on immunological/serological characteristics such as anti-neutrophil cytoplasmic antibodies (ANCA) or antibasement-membrane antibodies (ABMA) [1,
2,
4,
11].
I.
ANCA associated vasculitides refer to a group of heterogeneous autoimmune diseases that manifest with necrotising vasculitis and have positive anti-neutrophil antibody titres [4].
1.
Wegener’s granulomatosis (granulomatosis with polyangiitis)
Wegener’s is a systemic necrotising non-caseating granulomatous vasculitis [1-4].
The disease affects small to medium sized arteries,
capillaries,
and veins.
It initially affects the upper respiratory tract (sinuses and nasal mucosa) and then the lung parenchyma [3].
The lungs are affected in 90-100% of cases [1,
2,
4].
Additionally,
several other organs can also be affected - the kidneys,
pericardium,
skin,
eyes,
heart and joints [4,
5].
The strongest predilection is for the respiratory system and kidneys [4].
Renal involvement (necrotising glomerulonephritis) is usually secondary to pulmonary,
manifesting in 80-90% of cases [2,
4].
The classic triad of organ involvement encompasses the lungs,
upper respiratory tract/sinuses,
and kidneys.
Limited organ involvement of just the respiratory tract or especially just the kidneys is uncommon.
Widespread involvement includes systems outside of the classic triad [6].
The disease affects adults with a slight male predominance,
age margin of 30-40 years [2,
3].
Clinically,
granulomatosis with polyangiitis manifests with diffuse pulmonary haemorrhage,
cough,
and dyspnea.
Hemoptysis,
proteinuria and hematuria,
as well as systemic symptoms such as anorexia,
malaise and fever are also common [5].
Diffuse renal involvement may lead to acute renal failure [4].
Upper respiratory tract manifestations include mucosal ulcerations and granulomatous masses in the nasal cavities with damage to adjacent cartilage/bone [4].
Compared to Churg-Strauss syndrome,
Wegener’s clinical complex lacks a history of allergies or asthma [3].
The diagnosis of granulomatosis with polyangiitis is to be made on biopsy of involved tissues,
demonstrating the necrotising granulomatous inflammation and vasculitis characteristic of the disease.
The pathological changes in the renal parenchyma are often unspecific and biopsy may yield nondiagnostic results [2,
4].
High serologic titers for ANCA are specific for the diagnosis (90% of patients manifest these antibodies),
although a negative test does not exclude it [2,
4].
2.
Churg-Strauss syndrome (eosinophillic granulomatosis with polyangiitis/allergic angiitis and granulomatosis)
Churg-Strauss syndrome is a relatively rare ANCA-associated (positive in 75% of cases [4]) multisystem disorder.
It manifests with asthma,
blood eosinophilia (often dramatic),
necrotizing systemic vasculitis,
and extravascular granulomas [1-3,
8].
It involves small to medium arteries and veins.
Histologically Churg-Strauss syndrome is characterized by necrotizing vasculitis and extravascular granulomatous inflammation rich in eosinophils.
Hence it is also classified under the spectrum of eosinophilic lung disease [7].
Predominant involvement of just the small vessels is seldomly encountered,
therefore diffuse pulmonary haemorrhage is an uncommon manifestation of Churg–Strauss syndrome [1].
Additional clinical manifestations include mono- or polyneuropathy,
sinusitis,
history of atopic disease,
diarrhoea,
skin purpura,
and arthralgias [4,
8].
It is important to note that pulmonary involvement,
as seen radiographically or pathologically,
is indistinguishable from chronic eosinophilic pneumonia [2].
Additionally,
Churg-Strauss syndrome can be histologically identical to polyarteriitis nodosa or microscopic polyangiitis [4].
3.
Microscopic polyangiitis
Microscopic polyangiitis (MPA),
also known as “microscopic polyarteriitis nodosa”,
is a systemic autoimmune non-granulomatous necrotising vasculitis of small vessels.
It most often affects venules,
capillaries,
arterioles,
and small arteries,
although it occasionally involves medium-sized arteries [3,
9,
10].
MPA usually affects middle aged individuals with a median age of onset at 50 years [3,
9].
Some authors place it as the most common cause of pulmonary-renal syndrome [12].
It should be noted that microscopic polyangiitis and polyarteriitis nodosa are separate clinical entities – the latter affects medium-sized vessels only,
and pulmonary artery involvement and DAH are rare [9,
10].
The condition most commonly manifests necrotizing glomerulonephritis (present in 90-97% of cases [3,
9,
10]).
The lungs are affected by pulmonary capillaritis (25-50% of cases [9]) with relapsing DAH in 40% of cases.
Diffuse alveolar haemorrhage is evident in ~30% at presentation [10].
Purpura and gastrointestinal tract involvement may also occur.
It is uncommon for the eyes,
sinuses,
and upper airways to be affected.
MPA is ANCA-positive in ~70-90% of cases [4,
9].
Microscopic polyangiitis bears great histological similarity to polyarteriitis nodosa with the exception of involvement of smaller vessels – arterioles,
capillaries,
and venules (polyarteriitis nodosa only involves vessels larger than arterioles [4]).
MPA demonstrates absence or scarcity (paucity) of immunoglobulin deposition in vessel walls – this differentiates it from immune complex-mediated small vessel vasculitides (Schönlein-Henoch purpura and cryoglobulinaemic vasculitis) [4].
Some separate this condition into two organ specific subsets [4]:
Characteristically,
the clinical manifestation of MPA includes a long prodromal phase with symptoms such as fever and weight loss.
This is followed by the development of rapidly progressive glomerulonephritis [9].
Chest symptoms of microscopic polyangiitis include hemoptysis,
dry cough,
chest pain,
and shortness of breath progressing to irreversible air-flow obstruction [10].
II.
ANCA-negative vasculitis - Goodpasture’s syndrome
Goodpasture’s syndrome is an autoimmune disease,
characterised by damage to the alveolar and renal glomerular basement membranes induced by deposition of a cytotoxic antibody – antibasement-membrane antibody (ABMA).
It manifests the following triad:
-
pulmonary haemorrhage (DAH)
-
glomerulonephritis (RPGN)
-
circulating anti(glomerular-)basement membrane antibodies (ABMA) [4].
Although the age range is wide,
Goodpasture’s typically affects young adult caucasian men,
with women being affected three times less frequently [4,
9,
11].
Patients may present with cough,
dyspnoea,
haemoptysis and hypoxia.
Pulmonary manisfestations are usually the cause of presentation,
although most patients also have evidence of renal disease.
Usually glomerulonephritis develops after manifest haemoptysis/pulmonary involvement [9,
11].
Rarely,
the lung changes may isolated - without renal involvement [9].
The autoimmune reaction is with antibodies primarily directed against the collagen type IV molecules of renal glomerular basement membrane.
These antibodies (ABMA) also cross-react with alveolar basement membrane.
The diagnosis is made by immunofluorescent studies of renal or lung tissue,
which demonstrate the ABMA deposits as smooth wavy lines of fluorescent staining along the basement membrane [4,
9].
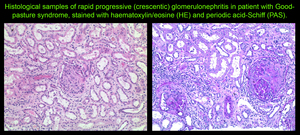
Fig. 2
References: M. Benkova, MD
It should be noted that Goodpasture’s manifests no other,
general vasculitic symptoms,
unlike the ANCA-associated small vessel vasculitides (Wegener’s,
Churg-Strauss,
MPA) [11].
III.
Systemic autoimmune conditions
1.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by inflammation,
deposition of immune complexes,
and vasculitis.
It can affect any age group,
but is most frequent in women (female to male ratio is reported between 9:1 and 13:1) between the ages of 20 and 40 [3,
4,
14].
The gender ratio is reversed in children,
with males being affected up to three times as often as females.
SLE involves different elements of the human immune system – the complement,
T-suppresor cells,
cytokine release,
with the end result being the production of freely circulating autoantibodies,
which may be present years before clinical manifestation [3,
4].
The disease tends to have a relapsing and remitting course.
SLE has a myriad of clinical features and radiographic presentations,
however,
in this article only those topically relevant will be discussed in detail - central nervous system,
gastointestinal,
and musculoskeletal manifestations are omitted.
Most of lupus patients arrive for imaging with an already clinically and immunologically establised diagnosis.
Imaging examinations are supplemental in diagnosing,
yet crucial in follow-up and detection of complications.
One of the primary characteristics of the disease is the presence of antiphospholipid syndrome (discussed as a separate entity further down) – responsible for the frequency and severity of cardiovascular and possibly renal involvement [3,
14].
It is accompanied by arterial and venous occlusive disease,
thrombocytopenia,
and recurrent vascular thrombosis.
Patients with antiphospholipid syndrome may present with pulmonary embolism,
pulmonary arterial hypertension,
and intraparenchymal pulmonary haemorrhage [3].
The respiratory system is often involved - in over half of SLE patients [1],
though the clinical manifestation is non-specific.
Equally common are damage to the pulmonary parenchyma,
to the pulmonary vessels,
and to the pleura and muscles of the chest wall [3,
4].
Pulmonary involvement and its imaging characteristics are discussed in depth in the following section.
A brief overview is provided in the following table:
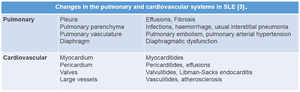
Table 1
References: Associate Professor G. Kirova, MD
Renal involvement is one of the more severe manifestations of lupus.
Most patients with SLE develop nephritis early in their disease course – usually within 5 years of diagnosis [14].
Lupus nephritis typically occurs in the same age group as the primary affected population – 20-40.
Children with SLE are at higher risk of renal disease than adults.
Men,
albeit suffering from SLE less frequently,
tend to develop lupus nephritis more frequently and have a poorer prognosis.
Lupus nephritis is histologically evident in most patients with SLE,
even those without clinical manifestations of renal disease.
The symptoms of lupus nephritis are generally related to hypertension,
proteinuria,
and renal failure.
The pathogenesis of lupus includes production of autoantibodies that form immune complexes intravascularly.
These complexes are then deposited in glomeruli.
Alternatively,
autoantibodies may bind to antigens already located in the glomerular basement membrane,
forming immune complexes in situ.
Immune complexes promote an inflammatory response by activating complement and attracting inflammatory cells.
Glomerular thrombosis is another mechanism that possibly partakes in the pathogenesis of lupus nephritis,
primarily in patients with antiphospholipid antibody syndrome.
It comprises an autoantibody attack against negatively charged phospholipid-protein complexes.
It is possible for lupus nephritis to be clinically silent,
however,
elevated serum creatinine levels,
low albumin levels,
or urinary protein or sediment suggest active nephritis [14].
Symptoms related to active nephritis may include peripheral edema secondary to hypertension or hypoalbuminemia.
Other symptoms directly related to hypertension include headache,
dizziness,
visual disturbances,
and signs of cardiac decompensation [14].
Differential diagnoses include:
-
Chronic Glomerulonephritis
-
Diffuse Proliferative Glomerulonephritis
-
Wegener Granulomatosis
-
Membranous Glomerulonephritis
-
Polyarteritis Nodosa
-
Rapidly Progressive Glomerulonephritis [14].
2.
Scleroderma (progressive systemic sclerosis)
Progressive systemic sclerosis (scleroderma) is a collagen vascular disease,
and one of the primary connective tissue diseases leading to irreversible damage to the pulmonary parenchyma [1-3].
It is characterized by deposition of excessive extracellular matrix proteins (mainly collagen) with following vascular occlusion due to gradual perivascular fibrosis.
It most frequently affects the joints (70-90%),
skin (hence scleroderma),
lungs (90%),
kidneys,
esopagus (80%),
and peripheral vasculature [1-4].
Similar to SLE,
progressive systemic sclerosis occurs more commonly in young and middle-aged women [1,
2].
Cutaneous features dominate the clinical picture initially [1].
The lungs are involved in 90% of patients,
yet only 25% manifest respiratory symptoms or plain radiographic evidence thereof [2,
4].
Pulmonary function testing is more sensitive than conventional radiography and shows the typical diminished lung volumes,
preserved flow rates,
and low diffusing capacity of interstitial pulmonary fibrosis [2].
Pulmonary vascular disease is the second most common finding in scleroderma.
Vasculitis is seen histologically as hyperplasia of the media and intima layers of small pulmonary vessels,
without accompanying severe pulmonary fibrosis.
Clinically vasculitis presents a diagnostic challenge until it manifests with intraparenchymal haemorrhage or secondary pulmonary arterial hypertension [3].
IV.
Antiphospholipid syndrome
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder.
It is usually defined as the clinical complex of recurrent vascular occlusion and ischaemic events,
and/or fetal loss,
occurring in patients who have circulating antiphospholipid antibodies [4,
15].
Multiple terms for APS exist,
some of which – misleading: for example,
lupus anticoagulant syndrome can be confusing since patients with antiphospholipid syndrome need not necessarily have SLE.
Some patients with APS do not manifest associated disease,
while in others the syndrome occurs in combination with lupus or another rheumatic/autoimmune disorder [15].
The currently preferred terminology is APS with/without associated disease; when associated,
APS was previously termed secondary.
APS with associated SLE may manifest lupus anticoagulant in addition to antiphospholipid antibodies [4].
The disease shows female predominance,
especially when associated with SLE.
It may affect all ages,
however,
there is a predilection for young to middle-aged adults.
Onset of antiphospholipid syndrome has been described in patients as young as 8 months.
[15]
The circulating antiphospholipid antibodies cross-react with cell membrane phospholipids.
This results in a hypercoagulable state leading to vascular thrombosis.
It can affect multiple organ systems [4,
15]:
-
obstetric (pregnancy loss,
eclampsia)
-
pulmonary (pulmonary thromboembolism,
pulmonary hypertension,
acute respiratory distress syndrome,
diffuse alveolar haemorrhage,
pulmonary capillaritis)
-
cardiac (Libman-Sacks valvulopathy,
myocardiac infarction)
-
renal (thrombotic microangiopathy)
-
peripheral venous system (deep venous thrombosis)
-
central nervous system (stroke,
sinus thrombosis,
seizures)
-
peripheral nervous system (peripheral neuropathy,
including Guillain–Barré syndrome).



