FINDINGS AND PROCEDURE DETAILS
As previously mentioned,
pulmonary-renal syndromes manifest with diffuse alveolar haemorrhage and glomerulonephritis.
The latter’s appearances on imaging vary with the evolution of the disease.
Often the only demonstrable abnormality is a change in size - always affecting both kidneys symmetrically.
In the acute phase the kidneys are enlarged due to swelling,
with no evident papillary/calyceal abnormality.
Later,
in the chronic phase,
the kidneys are small,
with smooth margins and normal pelvicalyceal systems.
The amount of renal sinus fat may appear excessive [1,
11].
Due to the relatively unspecific imaging findings of glomerulonephritis,
the diagnostic emphasis in regard to pulmonary-renal syndromes is on recognizing lung parenchyma changes,
demonstrated primarily by High Resolution Computed Tomography (HRCT).
Chest radiography has some diagnostic value as well,
though it is overshadowed by HRCT’s superior anatomical detail.
Magnetic resonance and ultrasound don’t provide insight into lung parenchymal involvement,
but are valuable in establishing manifest kidney changes,
as described above.
All herein presented CT imagery was yielded using a 64-slice dual-source multidetector scanner and/or a 2-slice dualdetector helical scanner.
Presented radiographs are all digitally acquired,
utilizing the digital radiography method with direct flat panel detection.
Diffuse Alveolar Haemorrhage (DAH)
Diffuse alveolar haemorrhage is a possible manifestation of all disease entities,
encompassed in this review.
It is their unifying pulmonary feature.
Radiographically,
DAH demonstrates as patchy,
usually extensive,
bilateral air-space opacities - consolidations and ground-glass.
Diffuse alveolar haemorrhage may be more prominent in the perihilar areas and in the middle and lower lung zones,
often sparing the lung apices and costophrenic angles [4,
10,
11].
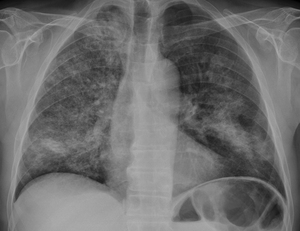
Fig. 3: Chest radiograph demonstrating bilateral patchy airspace opacities - primarily ground-glass. Patient was diagnosed with acute diffuse alveolar haemorrhage.
HRCT demonstrates a diverse pattern,
dependant on the time of onset of the haemorrhage.
It is imperative that this pattern be interpreted in light of clinical context.
In the acute phase the imaging findings can range from lobular/lobar ground-glass opacities to predominant consolidation.
Both of these patterns are due to alveolar filling with blood - partial in the case of ground-glass.
In contrast to the opacified airspaces,
segmental and subsegmental bronchi become prominent - air bronchograms (associated with consolidation) and “dark bronchus signs” - the latter being an identical phenomenon,
but associated with ground-glass [4,
10].
Within 2-3 days the ground-glass opacities may become superimposed by intralobular lines and smooth interlobular septal thickening.
These changes could even assemble into a crazy-paving pattern [13].
All changes in the acute/subacute phase may resolve completely [4].
In patients with chronic recurrent bleeding events ill-defined centrilobular nodules may become apparent.
These are due to intra-alveolar accumulation of haemosiderin-laden pulmonary macrophages.
The nodules are usually uniform in size and range between 1-3mm.
They are distributed diffusely and show no zonal predilection.
Severe repeated haemorrhage may progress to interstitial fibrosis [4].
I.
ANCA associated vasculitides
1.
Wegener’s granulomatosis (granulomatosis with polyangiitis)
Granulomatosis with polyangiitis can manifest with a multitude of radiographic appearances,
making diagnosis by imaging alone quite challenging at times.
Four pulmonary patterns are recognised,
although the first two are most common:
The most common radiological presentation (70% of cases [1]) is with multiple nodules (bilateral in 75% of cases [1]) of variable size randomly distributed throughout the lungs.
They range from well to poorly defined and may be distributed along bronchovascular bundles [1-4].
Nodules increase in size and number as the disease progresses [1].
In approximately half of cases,
some of the nodules demonstrate characteristic central cavitation at about 2 cm in size [1-4],
best demonstrated by CT.
Solitary nodules are reported in as many as a third of all patients [2].
Nodules commonly resolve spontaneously and reappear during relapse of the disease [3].
It should be emphasized that although the most common manifestation of recurrent/relapsing disease is cavitary nodules,
patients may relapse with a different pattern of involvement to their initial presentation [4].
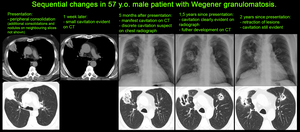
Fig. 4
References: M. Benkova, MD
Airspace consolidation and ground-glass opacities also may occur with or without the presence of nodules [1,
4].
Both nodules and regions of consolidation can have surrounding intraparenchymal haemorrhage,
demonstrated by poor peripheral demarcation,
and in some instances pulmonary haemorrhage dominates the presentation and radiographic appearance [3,
4].
Although a reticulonodular interstitial infiltrate at the bases is often the first manifestation,
it but usually asymptomatic.
In some cases focal patchy,
often peripheral regions of alveolar consolidation are seen,
which may also cavitate.
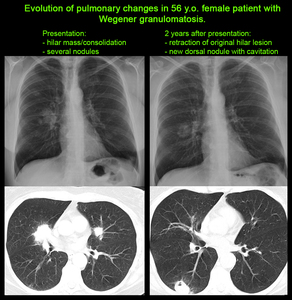
Fig. 5
References: M. Benkova, MD
Pleural effusions (unilateral or bilateral) are seen in 10-25% of cases,
usually as a result of cardiac or renal involvement – uncommon [1,
3].
Subglottic,
tracheal or bronchial thickening as well as calcified mucosal or submucosal deposits in the same zones may also be seen.
They produce irregular narrowing of the airway lumen - best demonstrated by CT [2].
Bronchial narrowing may result in lobar atelectasis [1].

Table 2
References: radiopaedia.org
2.
Churg-Strauss syndrome (eosinophillic granulomatosis with polyangiitis)
Plain radiography may demonstrate non-specific findings - peripheral transient consolidations and small pleural effusions (rarely) [3,
4].
Similarly,
HRCT imaging features are of low specificity - they largely reflect the eosinophilic infiltrate.
They include ground-glass opacities,
transient areas of poorly demarcated airspace consolidation of varying size - primarily peripheral and symmetrical [3]; centrilobular nodules (some of which may display cavitation),
and airways abnormalities attributable to asthma - bronchial wall thickening,
bronchiectasis,
and air-trapping [4].
Histologically the airspace disease is due to eosinophilic infiltrate or foci of organizing pneumonia.
Interlobular septal thickening may be seen as a result of interstitial pulmonary oedema secondary to cardiac involvement.
Pleural effusions and lymphadenopathy are uncommon [3].
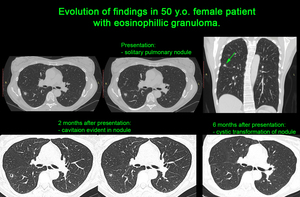
Fig. 6
References: Associate Professor B. Balev, MD
Unfortunately,
a significant proportion (up to 25%) of patients with Churg–Strauss syndrome have few or no imaging abnormalities and imaging is often of little help in establishing diagnosis - imaging diagnostic accuracy can be as low as 50% [1].
The transient consolidations may mimic chronic eosinophil pneumonia [3].

Table 3
References: radiopaedia.org
3.
Microscopic polyangiitis
The main radiologic feature is the image complex of diffuse alveolar haemorrhage (ground-glass and consolidation opacities),
which is seen in ~30-40% of patients [4,
9,
10].
Haemorrhages may relapse [4].
Additional (though non-specific) features include thickening of bronchovascular bundles,
centrilobular ground-glass nodules (indicating alveolar haemosiderin-laden macrophages) and diffuse honeycombing [4,
9,
10].
The latter is the primary feature of interstitial fibrosis – most often due to recurrent haemorrhage [9,
10].
Evidence of fibrosis may precede the onset of clinical symptoms by several years and is generally associated with the presence of serum ANCA.
Interstitial fibrosis can be a predictor for a poor prognosis [9].
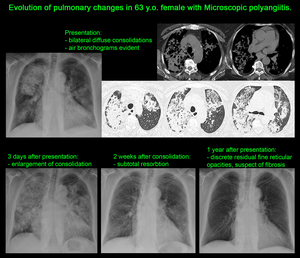
Fig. 7
References: M. Benkova, MD
Patients with a normal chest radiograph may demonstrate bilateral diffuse ground-glass opacities on HRCT [9].
Therefore HRCT is recommended in patients with clinically suspected pulmonary haemorrhage,
and especially in patients with acutely deteriorating renal function,
regardless of the presence of normal or ambiguous radiographic findings [9].
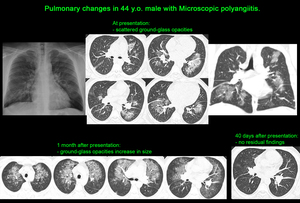
Fig. 8
References: M. Benkova, MD
The differential diagnosis can be quite broad – for pulmonary involvement it primarily includes Churg-Strauss syndrome and Wegener granulomatosis.
The former manifests with asthma and eosinophilia,
while the latter often has characteristic cavitating lesions [4].
II.
ANCA-negative vasculitis - Goodpasture’s syndrome
Plain radiography demonstrates non-specific changes.
Pulmonary haemorrhage (DAH) manifests as bilateral,
coalescent airspace opacities.
These resolve in several days to give a reticular pattern with the same distribution [4,
9].
Complete radiographic resolution is seen within 2-3 weeks.
CT findings are presented by ground-glass and consolidation opacities that progress to a reticular "crazy paving" pattern over a few weeks.
It should be noted that there is no interlobular septal thickening in the acute phase [4].
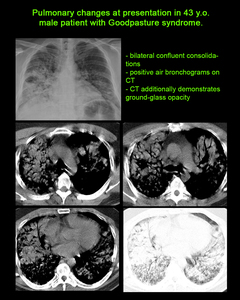
Fig. 9
References: M. Benkova, MD
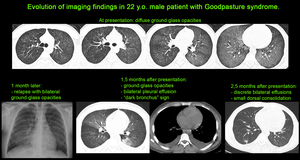
Fig. 10
References: M. Benkova, MD
Often radiographic and CT findings are indistinguishable from pulmonary oedema.
Other pulmonary vasculitides can also manifest similar imaging patterns – differentiation depends on pertinent serological and clinical data [4].
III.
Systemic autoimmune conditions
1.
Systemic lupus erythematosus
Pleural effusions are a manifestation of panserositis; they are a very frequent feature [1,
3,
4].
Usually they are bilateral,
small,
exudative in nature.
The presence of anti-DNA antibodies in pleural punctate is diagnostic and can differentiate systemic lupus manifestation from superimposed infection.
The most common pulmonary manifestations of SLE are lupus pneumonitis,
superimposed infections,
and intraparenchymal hemorrhage [3,
4].
Lupus pneumonitis has no exact single pathomorphological substrate as it comprises findings of:
-
non-specific interstitial pneumonia (HRCT - most commonly a combination of ground-glass and irregular linear/reticular opacities,
symmetrically or diffusely distributed in all zones or with basal predilection; possible subpleural sparing; scattered micronodules; traction bronchiectasis,
consolidation and honeycombing are late features [4]),
-
edema (secondary to renal/cardiac failure or capillaritis),
-
capillary damage and intraparenchymal haemorrhage [3].

Fig. 11
References: M. Benkova, MD
Intraparenchymal haemorrhages vary from episodic and latent to acute massive and lifethreatening.
In some cases they may be the first manifestation of the disease.
Radiographically,
SLE pneumonitis demonstrates uni- or bilateral zones of consolidation and ground-glass.
Development of respiratory distress syndrome is possible in more severe cases [3].
Cavitating nodules have also been described as a feature of pulmonary lupus involvement [4].
Pulmonary vascular disease in lupus encompasses pulmonary arterial hypertension,
intraparenchymal haemorrhage,
and pulmonary embolism.
Alveolar haemorrhage is relatively infrequent,
however,
when present,
it can have a severe clinical manifestation [3].
Conversely,
DAH may be the presenting feature of lupus [10].
It should also be stipulated that among the collagen vascular diseases,
SLE is the most common cause of DAH [10].
Conventional radiography demonstrates consolidations,
situated in the dependent parts of the lungs.
HRCT also readily identifies the pattern of DAH (described above); it is also very useful in controlling treatment and follow-up.
Pulmonary embolism is another vascular complication,
associated primarily with antiphospholipid syndrome.
The clinical and imaging constellation are typical,
with contrast enhanced CT being the method of choice to recognize thrombosis.
Cases with recurrent pulmonary embolisms develop arterial hypertension (demonstrable by perihilar pulmonary arterial dilatation and characteristic peripheral vascular “pruning”).
In contrast to scleroderma and rheumatoid arthritis,
SLE is not necessarily accompanied by marked pulmonary fibrotic changes.
HRCT is helpful when distinguishing between acute lupus pneumonitis and old fibrosis,
which can in some cases superimpose.
This is crucial in acute reversible disease,
where treatment can prevent complications.
The diaphragm is not to be overlooked when evaluating the chest changes of SLE.
Diaphragmatic dysfunction leads to suboptimal excursions and shrinking lung syndrome in up to 20% of patients [1-4].
Conventional radiography and CT alike show elevated hemidiaphragms with frequent basal flat (linear) atelectases [1-3].
2.
Scleroderma (progressive systemic sclerosis)
Pulmonary fibrotic changes are the primary finding in progressive systemic sclerosis.
The pulmonary parenchyma demonstrates the pattern of non-specific interstitial pneumonia [1-4],
which gradually progresses to irreversible fibrosis.
More rarely (in 5-10% of cases) scleroderma may manifest as usual interstitial pneumonia – reticular opacities in the immediate subpleural lung,
often associated with honeycombing and/or traction bronchiectasis,
with peripheral and lower lobe predominance; ground-glass opacities: usually less extensive than the reticular pattern and usually accompanying areas of reticulation or honeycombing; lung architectural distortion [1,
4].
Initially ground-glass opacities make up the only finding as a manifestation of active alveolitis.
Gradually permanent fibrosis settles in,
demonstrated by thickened interlobular septae and subpleural lines.
A typical feature is the involvement of the basal segments with development of traction bronchiectasis,
parenchymal desorganization,
and honeycombing.
As the disease progresses,
the entire parenchyma becomes affected.
In contrast to rheumatoid arthritis,
scleroderma patients manifest pleural and pericardial effusions much more seldomly.
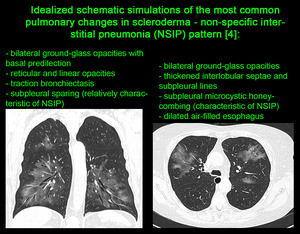
Fig. 12
References: G. Valchev, MD
Advanced pulmonary fibrosis in systemic sclerosis patients is a predisposing factor for the development of bronchoalveolar carcinoma [3,
4].
Finding spiculated foci against a background of reticular change mandates biopsy to exclude malignancy.
Pulmonary arterial hypertension in collagen disease has several pathogenetic mechanisms,
the primary among which are obliterative arteriitis,
microthromboembolism,
and progressive pulmonary fibrosis.
Pulmonary arterial hypertension is common in patients with scleroderma – over 10% develop the full clinical constellation during the course of the disease.
Plain film demonstrates dilated primary pulmonary artery branches with reduction (“pruning”) of peripheral vasculature.
HRCT demonstrates alternating zones of mosaic perfusion and normal lung parenchyma.
Pleural effusions are much less frequent in progressive systemic sclerosis than in other connective tissue diseases (rheumatoid disease and SLE) [2].
Pleural thickening is also rare and is demonstrated on HRCT in approximately 10% of patients.
It is more often attributable to extension of pulmonary interstitial fibrosis into the interstitial layer of the pleura than to pleuritis [2].
As in other diffuse fibrosing lung diseases,
enlarged (hyperplastic) mediastinal lymph nodes are a common feature on CT [1].
Eggshell calcification thereof has been reported,
although it is more common in silicosis and sarcoidosis [2].
Another helpful differential sign on CT is the presence of a dilated air-filled esophagus [4].
An air-fluid level therein suggests secondary distal esophageal stricture formation from chronic reflux esophagitis [2].
IV.
Antiphospholipid syndrome
Pulmonary involvement in antiphospholipid syndrome is one of its most frequent complications.
Arterial microthrombosis leads to a variety of conditions,
among which:
-
pulmonary thromboembolic disease
-
pulmonary arterial hypertension [4,
16]
-
acute respiratory distress syndrome
-
primary thrombosis of large lung vessels
-
microvascular pulmonary thrombosis
-
diffuse alveolar haemorrhage
-
fibrosing alveolitis
-
postpartum lung syndrome
-
pulmonary capillaritis [4].













